Cannabis Pharmacology
Abstract
The golden age of cannabis pharmacology began in the 1960s as Raphael Mechoulam and his colleagues in Israel isolated and synthesized cannabidiol, tetrahydrocannabinol, and other phytocannabinoids. Initially, THC garnered most research interest with sporadic attention to cannabidiol, which has only rekindled in the last 15 years through a demonstration of its remarkably versatile pharmacology and synergy with THC. Gradually a cognizance of the potential of other phytocannabinoids has developed. Contemporaneous assessment of cannabis pharmacology must be even far more inclusive. Medical and recreational consumers alike have long believed in unique attributes of certain cannabis chemovars despite their similarity in cannabinoid profiles.
This has focused additional research on the pharmacological contributions of mono- and sesquiterpenoids to the effects of cannabis flower preparations. Investigation reveals these aromatic compounds to contribute modulatory and therapeutic roles in the cannabis entourage far beyond expectations considering their modest concentrations in the plant. Synergistic relationships of the terpenoids to cannabinoids will be highlighted and include many complementary roles to boost therapeutic efficacy in treatment of pain, psychiatric disorders, cancer, and numerous other areas. Additional parts of the cannabis plant provide a wide and distinct variety of other compounds of pharmacological interest, including the triterpenoid friedelin from the roots, canniprene from the fan leaves, cannabisin from seed coats, and cannflavin A from seed sprouts. This chapter will explore the unique attributes of these agents and demonstrate how cannabis may yet fulfil its potential as Mechoulam’s professed “pharmacological treasure trove.”
ABBREVIATIONS
BCP beta-caryophyllene
CB1 cannabinoid type 1 receptor
CB2 cannabinoid type 2 receptor
DEA Drug Enforcement Agency
DEET N, N-dimethyl-toluamide
ECS endocannabinoid system
EO essential oil
FEMA Flavor and Extract Manufacturers’ Association
GRAS Generally Recognized As Safe
GVHD graft-vs-host-disease
MAGL monoacylglycerol lipase
MRSA methicillin-resistant Staphylococcus aureus
NAAA N-acylethanolamine-hydrolyzing acid amidase
PPARγ peroxisome proliferator-activated receptor gamma
TRP transient receptor potential
TRPA1 TRP ankyrin-type 1
1. INTRODUCTION
Mammals and plants are exposed to cannabinoids and related compounds that notably modulate their growth and physiology. The human species in the Old World grew up around the >70 million-year-old cannabis plant, giving us a natural affinity to cannabinoids (Clarke & Merlin, 2012). This plant has been documented as a provider of food, clothing, textiles, and medicine for millennia. For thousands of years, the plant has been associated with relieving symptoms of disease and has demonstrated numerous therapeutic properties (Russo, 2007, 2011). In this century, we are finally beginning to understand the precise pharmacological mechanisms underlying the effects of cannabis and related preparations, most of which can be explained through the endocannabinoid system (ECS). As perhaps the most significant human biological scientific discovery in the last 30 years, the ECS is only now being integrated into medical school curricula.
Analytical chemistry has revealed a rich and abundant “pharmacological treasure trove” in the plant. Compounds that may affect the pharmacology of cannabinoids are abundant in nature, and so we may dangerously and mistakenly consider their presence to be trivial. If so, this could cause us to lose sight of the subtlety and efficiency of their design when applied in combination. There are some 100 clinical studies and thousands of articles on the pharmacology and pharmacodynamics of cannabis and its influence on how humans eat, sleep, heal, and learn. In this review, we hope to demystify some of the wonder of cannabis as a medicine by providing a concise overview of the pharmacological mechanisms of cannabis compounds, which will hopefully guide medical school curricula, advances in therapies, and lead to changes in public health approaches both nationally and internationally. As government information sources are updated with cannabis research conducted in the current century, the future of cannabis in society will depend strongly on how well we understand this plant, of which our access to for research and medicine currently floats on the winds of politics (Fig. 1).
2. CANNABIS PHYTOCANNABINOIDS
2.1 Tetrahydrocannabinol
The pharmacology of tetrahydrocannabinol (THC) is perhaps the most well studied of any scheduled substance, having well over 100 published clinical studies of medical cannabis and related products which contain THC (Ben Amar, 2006; Hazekamp & Grotenhermen, 2010; Kowal, Hazekamp, & Grotenhermen, 2016; Marcu, 2016; Pertwee & Cascio, 2014; Russo & Hohmann, 2012). THC, among a pantheon of over 100 (Hanus, Meyer, Munoz, Taglialatela-Scafati, & Appendino, 2016), is the most common phytocannabinoid in cannabis drug chemotypes, and is produced in the plant via an allele codominant with CBD (de Meijer et al., 2003). THC displays both cannabinoid receptor-dependent and independent mechanisms. THC interacts efficiently with CB1 (Ki ¼5.05–80.3 nM) and CB2 receptors (Ki ¼1.73–75.3 nM), which underlies its activities in modulating pain, spasticity, sedation, appetite, and mood (Russo, 2011). Additionally, it is a bronchodilator (Williams, Hartley, & Graham, 1976), neuroprotective antioxidant (Hampson, Grimaldi, Axelrod, & Wink, 1998), antipruritic agent in cholestatic jaundice (Neff et al., 2002) and has 20 times the antiinflammatory power of aspirin and twice that of hydrocortisone (Evans, 1991).

Fig. 1 The cannabis plant, its parts, and their phytochemical components. Component percentages are based on information from Callaway
(2004), (Meier & Mediavilla, 1998) and Potter (2009) (all photos by EBR).
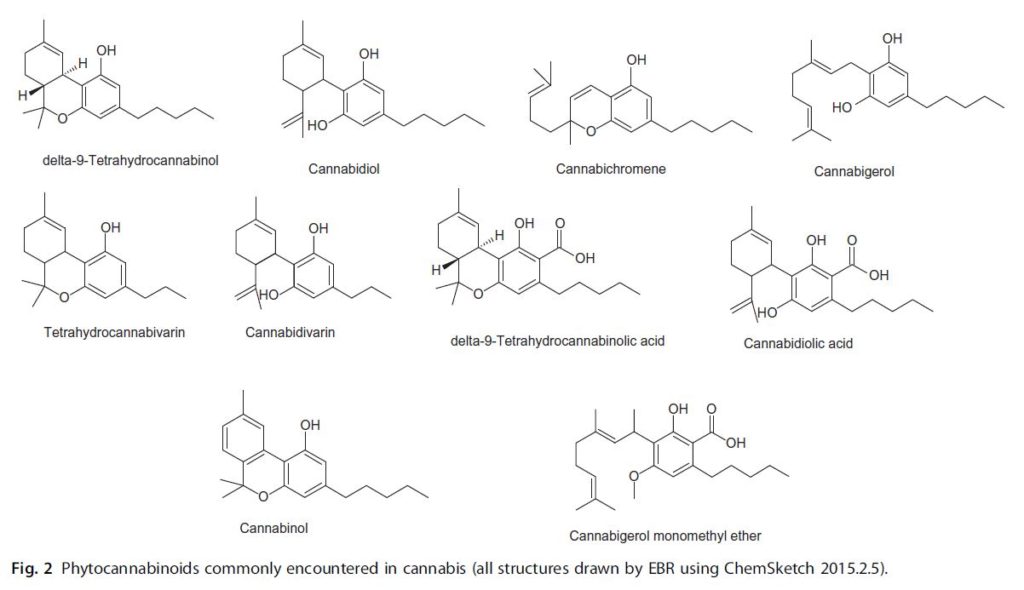
Fig. 2 Phytocannabinoids commonly encountered in cannabis (all structures drawn by EBR using ChemSketch 2015.2.5)
THC is likely to avoid potential pitfalls of either COX-1or COX-2 inhibition, as such activity is only noted at concentrations far above those attained therapeutically (Stott, Guy, Wright, & Whittle, 2005). While THC stimulates both CB1 and CB2 receptors, the role and distribution of these two proteins is distinct. Stimulation of CB1 receptors by THC can lead to a tetrad of effects in assays with laboratory animals; these effects include: suppression of locomotor activity, hypothermia, catalepsy (ring test), and antinociceptive effects in the tail flick test (Martin et al., 1991). CB2 receptor stimulation is associated with pain relief and anti-inflammatory activities (Pacher & Mechoulam, 2011), but it is not associated with other CB1 effects such as appetite stimulation.
2.1.1 THC Mechanisms at CB1 and CB2
THC-mediated CB1 receptor stimulation inhibits forskolin-stimulated adenylate cyclase (AC) and leads to the inhibition N-, Q-, L-type calcium channels. Ion channels can be modulated from CB1 receptor stimulation. For example, CB1 receptor stimulation releases G proteins to activate inwardly rectifying potassium channels, which may be induced by a variety of CB1 partial agonists (Console-Bram, Marcu, & Abood, 2012). This receptor signaling also stimulates the activity of MAP kinases. MAP kinase pathways are often activated by GPCRs and can alter the activity of ERK1/2, c-Jun N-terminal kinase (JNK), p38 MAP kinase, and/or ERK5 proteins. The stimulation of their activity can control cell growth and their metabolism. CB1 localization is widespread, and the distribution parallels the known pharmacological actions of THC; the locations of CB1 receptors make them a good therapeutic target (Herkenham et al., 1990; Pacher, 2006; Russo, 2016a). CB1 has particularly high expression in neuronal tissue, specifically in pre- and postsynaptic neurons in the central nervous system (CNS). CB1 protein is found in the nucleus of solitary tract (i.e., antiemetic effects), hypothalamus, motor systems, motor cortex, basal ganglia, cerebellum, spinal cord (motor neurons in spinal cord), eye, sympathetic ganglia (also enteric nervous system), immune system (bone marrow, thymus, spleen, tonsils), breast cancer cell lines, and other peripheral sites such as the heart, lungs, adrenals, kidneys, liver, colon, prostrate pancreas, testes, ovaries, and placenta. THC-mediated CB2 receptor stimulation leads to inhibition of forskolin-stimulated AC activation and stimulating MAP kinases but lack the effects on ion channels of CB1. CB2 is localized mainly in cells of the immune system, such as bone marrow, thymus, spleen, tonsils, T and B lymphocytes, monocytes, NK cells, PMN, and mast cells. The levels of CB2 expression increase during activation/differentiation of immune cells. During inflammation or injury, the number of CB2 receptors available for stimulation increases significantly. CB2 is also found in tissue of the uterus, lung, bone (osteoclasts, osteoblasts, and osteocytes), microglia, and brainstem neurons. CB2 DNA mutations or polymorphisms are associated with osteoporosis in human populations, and strains of mice that are engineered without CB2 can have accelerated age-related trabecular bone loss. The maximal effect of THC at the CB receptor proteins is well below that of synthetic cannabinoids (i.e., nabilone, HU-210, JWH-018, etc.). Hence, THC, as well as anandamide, are classified as partial agonists because other ligands or cannabinoids exist, which are much more potent at cannabinoid receptors (Matsuda, Lolait, Brownstein, Young, & Bonner, 1990; Pacher, 2006). For example, 11-hydroxy metabolites of THC that are generated by the liver from oral administration of THC interact more efficiently at CB1 receptors. It should also be noted that cannabinoid drugs with equal (i.e., Marinol®) or greater (i.e., nabilone) potency than THC, have been approved and available by prescription for decades, but no significant black market exists for these expensive and hard to obtain standardized preparations nor has addiction treatment been a significant issue for these cannabis-based medicines (Calhoun, Galloway, & Smith, 1998; Robson, 2011).
2.1.2 THC Activity Independent of CB1 and CB2
THC has been reported to interact with a wide variety of proteins including various receptors, channels, and enzymes. These pharmacological actions of THC are well documented in biochemical and mammalian research studies. Findings and research demonstrating actions of THC above 10 μMconcentration are beyond the scope of this chapter as beyond this concentration, the results become difficult to interpret as far as what the physiological significance could be.
2.1.3 Receptors and Channels
At <1 μM THC can activate GPR18, GPR55, peroxisome proliferator activated receptor gamma (PPARγ) nuclear receptors, as well as TRPA1 and TRPV2 cation channels, while enhancing the activity of non-CB receptors on sensory neurons mediating the release of calcitonin gene-related peptide (an effector in migraine attacks) and potentiating glycine-ligated ion channels (important for pain relief ) (Hong & Liu, 2017). Conversely, THC blocks or antagonizes the activity of 5-HT3A ligand-gated ion andTRPM8 cation channel at <1 μM. Between 1 and 10 μM, THC can activate the PPARγ nuclear receptor, TRPV3 and TRPV4 cation channels, and potentiate the activity of β-adrenoceptors. THC can either block or activate GPR55 at these concentrations, depending on experimental conditions. Perhaps most relevant to current clinic and public health issues is the ability of THC to displace opiates from the μ-opioid receptor, as well as allosterically modulate the μ- and δ-opioid receptor to inhibit their activity between 1 and 10 μM (Lichtman, Sheikh, Loh, & Martin, 2001; Pertwee et al., 2010). This perhaps underlies the potential of cannabis as part of a viable solution to the opiate crisis in terms of treating addiction, withdrawal, and harnessing the benefits of cannabinoid-opiate coadministration in the clinic (Americans for Safe Access, 2016). When THC and morphine are coadministered, ¼ the dose of morphine is required to reach significant reductions in pain (Naef et al., 2003). Conversely, THC inhibits T-type calcium (Cav3) voltage gated ion channels, potassium Kv1.2 voltage-gated ion channels, conductance in Na+ voltage-gated ion channels (), and conductance in gap junctions between cells at concentrations between 1 and 10 μM. THC can also interact with a variety of enzymes such as phosphlipases, lysophosphatidylcholine acyl transferase, lipoxygenase, Na+-K+-ATPase, Mg2+-ATPase, CYP1A1, CYP1A2, CYP1B1, CYP2B6, CYP2C9, and monoamine oxidase activity (Evans, 1991; Pertwee, 1988; Pertwee & Cascio, 2014; Yamaori et al., 2012; Yamaori, Kushihara, Yamamoto, & Watanabe, 2010; Yamaori, Okamoto, Yamamoto, & Watanabe, 2011). The synaptic conversion of tyrosine to noradrenaline and dopamine (DA) is increased by THC while norepinephrine-induced melatonin biosynthesis is inhibited. Recently, THC has shown significant benefits in helping to reduce complications during organ transplant and in graft-vs-host-disease (GVHD) in mammals. The research on THC in GVHD and transplant has already affected public policy in California, where cannabis use no longer constitutes grounds for being dismissed from transplant waiting list. The perceived pharmacological effects of THC may also be dependent on diet of the mammal (Balvers et al., 2012; Lafourcade et al., 2011; Lowette, Roosen, Tack, & Berghe, 2015), due to the fact that anandamide and endocannabinoids are derived in vivo from omega-3 and -6 fatty acid intake and their dietary deficiency could lead to uncoupling of G protein-coupled receptors.
2.2 Cannabidiol
The main nonintoxicating phytocannabinoids are cannabidiol (CBD) and its acidic precursor cannabidiolic acid. These are the most abundant phytocannabinoids in European hemp (Upton et al., 2013). CBD has a very low affinity for CB receptors but may have significant CB1- and CB2-independent mechanisms of action and possess the unique ability to antagonize CB1 at very low concentrations when in the presence of THC (Thomas et al., 2007). This observed antagonism may be related to CBD’s ability to act as a negative allosteric modulator at CB1 receptors (Laprairie, Bagher, Kelly, & Denovan-Wright, 2015).
CBD is reported to be an agonist at TRPV1 (Bisogno et al., 2001) and 5-HT1A receptors (Russo, Burnett, Hall, & Parker, 2005) and to enhance adenosine receptor signaling (Carrier, Auchampach, & Hillard, 2006). Exceptional tolerability of CBD in humans has been demonstrated (Mechoulam, Parker, & Gallily, 2002). CBD can produce a wide range of pharmacological activity including anticonvulsive, antiinflammatory, antioxidant, and antipsychotic effects. These effects underlie the neuroprotective properties of CBD and support its role in the treatment of a number of neurological and neurodegenerative disorders, including epilepsy, Parkinson disease, amyotrophic lateral sclerosis, Huntington disease, Alzheimer disease, and multiple sclerosis (de Lago & Ferna´ndez-Ruiz, 2007; Hofmann & Frazier, 2013; Martin-Moreno et al., 2011; Scuderi et al., 2009).
CBD possesses the unique ability to counteract the intoxicating and adverse effects of cannabis, such as anxiety, tachycardia, hunger, and sedation in rats and humans (Murillo-Rodriguez, Millan-Aldaco, Palomero-Rivero, Mechoulam, & Drucker-Colin, 2006; Nicholson, Turner, Stone, & Robson, 2004; Russo, 2011; Russo & Guy, 2006). The benefits of CBD include reducing the unwanted side effects of THC, a dynamic pharmacological effect that has been fairly well studied in clinical trials. CBD is included in a specific ratio of 1:1 in the medicinal cannabis preparation and licensed pharmaceutical known as Sativex®, which has been studied in numerous properly controlled clinical trials representing thousands of patient/years of data (Flachenecker, Henze, & Zettl, 2014; Rog, Nurmiko, Friede, & Young, 2005; Sastre-Garriga, Vila, Clissold, & Montalban, 2011; Wade, Collin, Stott, & Duncombe, 2010).
Recently, CBD demonstrated its strong antiinflammatory and immunosuppressive properties in a phase II study on GVHD (Yeshurun et al., 2015). CBD (300 mg/day) starting a week before the procedure was associated with less mortality and complications.
There is recent report that CBD isomerizes to THC under acidic conditions in vitro, but there is no evidence that directly supports that this is actually occurring in humans (Deiana et al., 2012; Grotenhermen, Russo, & Zuardi, 2017; Russo, 2017).
2.3 Cannabigerol
This compound was purified from cannabis the same year as THC (Gaoni & Mechoulam, 1964), but cannabigerol (CBG) lacks its psychotropic effects (Grunfeld & Gresty, 1998; Grunfeld & Edery, 1969).Normally, CBG appears as a relatively low concentration intermediate in the plant, but recent breeding work has yielded cannabis chemotypes lacking in downstream enzymes that express 100% of their phytocannabinoid content as CBG (de Meijer & Hammond, 2005; de Meijer, Hammond, & Micheler, 2009). CBG, the parent phytocannabinoid compound, has a relatively weak partial agonistic effect at CB1 (Ki 440 nM) and CB2 (Ki 337 nM) (Gauson et al., 2007).
CBG may stimulate a range of receptors important for pain, inflammation, and heat sensitization. This compound can antagonize TRPV8 receptors and stimulates TRPV1, TRPV2, TRPA1, TRPV3, TRPV4, and α2-adrenoceptor activity (Cascio, Gauson, Stevenson, Ross, & Pertwee, 2010; De Petrocellis & Di Marzo, 2010; De Petrocellis et al., 2011). It is a relatively potent TRPM8 antagonist for possible application in prostate cancer and detrusor overactivity and bladder pain (De Petrocellis & Di Marzo, 2010; Mukerji et al., 2006). CBG can also antagonize the stimulation of serotonin 5-HT1A and CB1 receptors with significant efficiency. Older work supports gamma aminobutyric acid (GABA) uptake inhibition greater than THC or CBD that could suggest muscle relaxant properties (Banerjee, Snyder, & Mechoulam, 1975).
Analgesic and antierythemic effects and the ability to block lipooxygenase were said to surpass those of THC (Evans, 1991). CBG demonstrated modest antifungal effects (ElSohly, Turner, Clark, & Eisohly, 1982). CBG has remarkable anticancer properties in basic research models, it has proved to be an effective cytotoxic in high dosage on human epithelioid carcinoma and is one of the more effective phytocannabinoids against breast cancer (Baek et al., 1998; Ligresti et al., 2006). CBG has significant antidepressant effects in the rodent tail suspension model and is a mildly antihypertensive agent (Maor, Gallily, & Mechoulam, 2006; Musty & Deyo,
2006).
Additionally, CBG inhibits keratinocyte proliferation suggesting utility in psoriasis (Wilkinson & Williamson, 2007). CBG is a strong AEA uptake inhibitor and a powerful agent against MRSA (methicillin-resistant Staphylococcus aureus) (Appendino et al., 2008; De Petrocellis et al., 2011). Finally, CBG behaves as a potent α2- adrenoreceptor agonist, supporting analgesic effects previously noted, and moderate 5-HT1A antagonist suggesting antidepressant properties (Cascio et al., 2010; Formukong, Evans, & Evans, 1988).
2.4 Cannabichromene
Cannabichromene (CBC) was first reported to be isolated by two groups, using either a hexane/florisil extraction method from hashish or a benzene percolation of hemp (Claussen, Von Spulak, & Korte, 1966; Gaoni & Mechoulam, 1966). This cannabinoid represents 0.3% of constituents from confiscated cannabis, and it is important to note that varieties and preparations exist in the commercial and medical markets with significantly higher content (de Meijer & Limited, 2011; Mehmedic et al., 2010; Meijer, Hammond, & Micheler, 2008; Swift, Wong, Li, Arnold, & McGregor, 2013). CBC-rich cannabis strains are the result of selecting for the inheritance of a recessive gene, achievable through extensivecross-breeding. CBC or CBC-like derivatives have also been found in Rhododendron anthopogonoides, at the time of this writing, this species and its extracts are not listed under the list of scheduled drugs by the DEA (Iwata & Kitanaka, 2011).
CBC can interact with transient receptor potential (TRP) cation channels that inhibit endocannabinoid inactivation, and stimulate CB2 receptors (Ki 100 nm), but it does not have significant activity at CB1 receptors (Ki>1 μM) (De Petrocellis et al., 2011, 2012, 2008; Shinjyo & Di
Marzo, 2013). TRP channels and the ECS are involved in inflammation and have a role in pain. In mice, CBC can relieve pain, potentiate the analgesic effects of THC, ameliorate-induced colonic inflammation, and paw edema by demonstrably inhibiting macrophage and MAGL activity (Cascio & Pertwee, 2014; Davis & Hatoum, 1983; Maione et al., 2011).
The mechanism underlying CBC’s observed effects in mammals is supported by pharmacodynamic studies (De Petrocellis et al., 2008; Ligresti et al., 2006; Romano et al., 2013). These have shown that CBC can stimulate TRP ankyrin-type 1 (TRPA1) cation channels (EC50 ¼90 nM), and desensitize these channels (IC50 ¼370 nM). Further evidence for the role of CBC in inflammation includes the compounds ability to interact with TRPV4 and TRPV3 cation channels (EC50 ¼600 nM and 1.9 μM, respectively), and desensitize TRPV2 and TRPV4 (IC50 ¼6.5 and 9.9 μM, respectively) (Cascio & Pertwee, 2014; De Petrocellis et al., 2012).
Beyond inflammation and pain, CBC may have a positive effect on the viability of mammalian adult neural stem cell progenitor cells, which are an essential component of brain function in health and disease (Shinjyo & Di Marzo, 2013). In summary, CBC can be one of the most abundant nonintoxicating CBs found in cannabis, due to a recessive gene (Brown & Harvey, 1990;
Holley, Hadley, & Turner, 1975). CBC can cause strong anti-inflammatory effects in animal models of edema through non-CB receptor mechanisms (DeLong, Wolf, Poklis, & Lichtman, 2010). CBC has been shown to significantly interact with TRP cation channels, including TRPA1, TRPV1–4, and TRPV8 (Pertwee & Cascio, 2014). CBC can also produce behavioral activity of the cannabinoid tetrad. The effects of CBC, particularly nociception in animal models, can be augmented for additive results when THC is co-administered.
2.5 Cannabinol
Cannabinol (CBN) is the nonenzymatic oxidation byproduct of THC and is most commonly an artifact found after prolonged storage, especially at higher temperatures. CBN was the first cannabinoid to be identified and isolated from cannabis (Wood, Spivey, & Easterfield, 1899). This discovery was most likely due to rampant degradation of THC to CBN due to poor quality control, the transportation and storage conditions related to the 19th century; challenges that are still difficult to overcome in existing cannabis products (Upton et al., 2013).
Relative to THC, CBN maintains about ¼ the potency (Ki at CB1 ¼211.2 nM, CB2 ¼126.4 nM) (Rhee et al., 1997). CBN can be sedative, anticonvulsant in animal and human studies, and has demonstrated significant properties related to antiinflammatory, antibiotic, and anti-MRSA activity (minimum inhibitory concentration (MIC) 11 μg/mL) (Appendino et al., 2008; Evans, 2007; McPartland & Russo, 2001; Musty, Karniol, Shirikawa, Takahashi, & Knobel, 1976; Turner, Elsohly, & Boeren, 1980).
CBN has potential as a component in topical applications, inhibiting keratinocyte proliferation (low micromolar) via CBR-independent mechanisms, suggesting utility in psoriasis (Wilkinson & Williamson, 2007). Beyond cannabinoid proteins, the compound has TRPV2 (high-threshold thermosensor) agonistic effects (EC50 77.7 μM), which are of interest in possible topical applications in treating burns (Qin et al., 2008; Russo, 2014). A review of phytocannabinoids summarized the ability of CBN to inhibit the activity of a number of enzymes, including cyclooxygenase, lipoxygenase, and a host of cytochrome P450 (CYP) enzymes (e.g., CYP1A1, CYP1A2, CYP2B6, CYP2C9, CYP3A4, CYP3A5, CYP2A6, CYP2D6, CYP1B1, and CYP3A7) (Pertwee&Cascio, 2014). CBNmay also stimulate the activity of phospholipases. CBNadditionally stimulates recruitment of quiescentmesenchymal stem cells in marrow (10 μM) promoting bone formation (Scutt & Williamson, 2007) and can affect breast cancer resistance proteins (IC50 approximately 145 μM) (Holland, Allen, & Arnold, 2008).
2.6 Tetrahydrocannabivarin
Tetrahydrocannabivarin (THCV) is a propyl analogue of THC most often encountered in low concentration in dried plant material, but in THCVrich plants up to 16% THCV by dry weight has been recorded (Meijer & Hammond, 2005). Mechanistically speaking, THCV can behave as both an agonist and an antagonist at CB1 receptors depending on the concentration (Pertwee, 2008). THCV produces weight loss, and decreases body fat and serum leptin concentrations with increased energy expenditure in obese mice (Cawthorne, Wargent, Zaibi, Stott, & Wright, 2007; Riedel et al 2009). THCV also demonstrates prominent anticonvulsant properties in rodent cerebellum and pyriform cortex (Hill et al., 2010). THCV appears as a fractional component of many southern African cannabis chemotypes, although plants highly predominant in this agent have been produced (de Meijer et al., 2003; de Meijer & Hammond, 2016). THCV has the CB2- based ability to suppress carageenan-induced hyperalgesia and inflammation, and both phases of formalin-induced pain behavior via CB1 and CB2 in mice (Bolognini et al., 2010).
Antagonizing CB1 receptors can suppress appetite and the intoxicating effects of THC.
However, caution must be emphasized when developing CB1 receptor antagonists. Clinical studies in human populations studying the antagonists of CB1 receptors with the drug rimonabant (SR141716A) led to depressive episodes and potentially worsened neurodegenerative disease outcomes, and ultimately this drug was withdrawn from the market (McLaughlin, 2012). Despite this setback, SR141716A remains a very important research tool for unlocking potential medical treatments targeting the CB receptors and deepening the understanding of the ECS. Importantly, the neutral antagonism mechanism of action of THCV seems to be free of the adverse events associated with the CB1 inverse agonists (McPartland, Duncan, Di Marzo, & Pertwee, 2015).
2.7 Tetrahydrocannabinolic Acid
Cannabinoid acids are found as primary metabolites in cannabis plants. For example, tetrahydrocannabinol acid (THCA-A) is synthesized in glandular trichomes of the cannabis plant and forms THC after the parent compound is decarboxylated by UV exposure, prolonged storage, or heat (Moreno- Sanz, 2016). THCA-A can represent up to 90% of total THC content in the plant, it has about 70% conversion rate into THC when smoked (Dussy, Hamberg, Luginbuhl, Schwerzmann, & Briellmann, 2005): decarboxylation of THCA to THC is incomplete even at high temperatures in gas chromatography. Additionally, THCA can be detected in serum, urine, and oral fluid of cannabis consumers up to 8 h after smoking (Jung, Kempf, Mahler, & Weinmann, 2007). The cannabinoid acids do not produce any significant or documented psychotropic effects. THCA-A is the immediate natural precursors of THC. THCA-A is one the primary phytocannabinoid metabolites and can cause apoptosis of insect cells (Sirikantaramas et al., 2004).
THCA-A is reported to be a weak agonist of CB1 and CB2 receptors compared with THC (Ki CB1¼630 vs 3.5 nM; Ki CB2 ¼890 vs 3.2 nM) (Verhoeckx et al., 2006). In other laboratories, THCA-A effectively bound to both cannabinoid receptors, displaying a higher affinity for CB1, with Ki values of 23.51–3.5 and 56.13–8.2 nM, respectively. In fact, THCA-A (log IC50 ¼1.793 0.00) and THC (log IC50 ¼1.941–0.01) displaced CP-55,940 from CB1 in a similar range of concentrations (Rosenthaler et al., 2014).
THCA-A attenuated nausea-induced gaping in rats and vomiting in shrews through a mechanism that required CB1 activation, which is reversible with a CB1 receptor antagonist (Rock, Kopstick, Limebeer, & Parker, 2013). The authors provide additional evidence that this observed effect of THCA-A is not due to the conversion of THCA-A to THC. The effects of THCA-A appear to be partially mediated through cannabinoid receptors, without any reported psychotropic effects associated with THC. The evidence suggests that THCA-A is restricted to the periphery with limited access to the CNS through the blood brain barrier (BBB). This is probably due the presence of a carboxylic acid on THCA-A; such polar residues decrease CNS penetration through the ATP-binding cassette family of transporters (Moreno-Sanz et al., 2013). In fact, brain disposition has been reported for several cannabinoids, but not THCA-A (Alozie, Martin, Harris, & Dewey, 1980; Deiana et al., 2012).
THCA-A can inhibit the release of tumor necrosis factor-alpha (TNF-α) (Verhoeckx et al., 2006), can efficiently interact with TRPM8 channels and can stimulate or desensitize a range of other TRP cation channels. THCA-A has been found to inhibit enzymes responsible for the breakdown of endocannabinoids, as well as COX-1 and -2, thus stimulating the ECS by increasing levels of endogenous cannabinoids. In a basicmodel of Parkinson’s disease, THCA-A (10 μM) increased cell survival and significantly ameliorated altered neurite morphology (Moldzio et al., 2012). THCA-A reduces cell viability of various cancer cell lines when administered in vitro (Moreno-Sanz, 2016). Basic research has conclusively shown that THCA-A can have immunomodulatory, anti-inflammatory, neuroprotective and antineoplastic activity.
2.8 Cannabidivarin
Cannabidivarin (CBDV) was probably first reported in a benzene extract from a Thai cannabis variety referred to as “Meao” (Shoyama, Hirano, Makino,Umekita,&Nishioka, 1977). Oral CBDV (60 mg/kg) administered to rats can cross the BBB (Deiana et al., 2012). CBDV is capable of activating and blocking, depending on experimental conditions, a diverse number of cation channels. At less than <1 μM TRPA1, TRPM8, and TRPV4 are influenced by CBDV, while around 1–10 μMaffects the activity of TRVP1, TRVP2, and TRVP3 cation channels. In addition to cationic influences, this propyl analogue of CBD engages the ECS by inhibiting endocannabinoid degradation at 10 μM through modulating the rate of diacylglycerol lipase activity and N-acylethanolamine-hydrolyzing acid amidase (NAAA), the effects of which could magnified with CBDV’s ability to inhibit the cellular uptake of anandamide (Pertwee & Cascio, 2014). CBDV also possess the potential for the treatment of nausea and vomiting (Rock, Sticht, & Parker, 2014). There is strong evidence that CBDV has significant anticonvulsant properties, which may rival CBD’s therapeutic potential in treating epilepsy, particularly seizures of partial onset (focal seizures) (Williams, Jones, & Whalley, 2014).
2.9 Cannabidiolic Acid
Cannabidiolic acid (CBDA) is the natural precursor or CBD, and this acidic phytocannabinoid can targetGPR55, TRPA1, TRPV1, and TRPM8 at concentrations between 1 and 10 μM. At higher concentrations, the compound can inhibit ECS degradation enzymes. CBDA can inhibit by COX 1 and COX-2 (Takeda, Misawa, Yamamoto, & Watanabe, 2008). CBDA also shares CBD’s ability to enhance 5-HT1A receptor activation but the acidic compound does not interact efficiency with CB1 receptors as either an agonist or antagonist (Bolognini et al., 2013; McPartland et al., 2015). The affinity at 5-HT1A for CBDA is greater than an order of magnitude higher compared to CBD. Evidence from animals demonstrates significant antiemetic effects from CBDA (10 or 200 mg/kg ip) (Moreno-Sanz, 2016; Rock & Parker, 2015).
2.10 Cannabigerol Monomethyl Ether
This phytocannabinoid is commonly encountered in cannabis, but has not been researched for pharmacological activity. It is included here to highlight that its presence with relative frequency supports its investigation as a research priority.
3. CANNABIS TERPENOIDS
Terpenoids are aromatic compounds that fulfill unique ecological roles for plants in protection from predation, attraction of pollinators, and myriad other roles (Elzinga, Fischedick, Podkolinski, & Raber, 2015; Fischedick, Hazekamp, Erkelens, Choi & Verpoorte, 2010; McPartland & Russo, 2001, 2014; Russo, 2011). Two excellent general references are Baser and Buchbauer (2016) and Langenheim (1994). They are typically produced in dedicated structures,which in the case of cannabis are the glandular trichomes, the same source of phytocannabinoid production (Potter, 2009). Typically, many are produced by a given plant and form its essential oil (EO). In cannabis, the biochemical diversity of these components is remarkable, with as many as 200 described, although some are artifacts of steam distillation (Lawless, 1995).
The biochemical profile of terpenoids in a given plant ismore genetically than environmentally determined (Franz & Novak, 2010). Whereas, the biosynthetic enzymes for phytocannabinoids have been identified for several years, it was only recently that several terpenoid synthases were analyzed in cannabis (Booth, Page & Bohlmann, 2017). Regulation of terpenoid and cannabinoid production in the plant remain important research priorities. A great deal of debate has surrounded the relative importance, or lack thereof, of cannabis terpenoids to the pharmacological effects of the plant. Despite existing at seemingly low concentrations in a preparation, they have proven to be potent: small amounts in ambient air produce marked behavioral effects to increase or decrease activity levels in rodents, even when observed serum levels are low or negligible (Buchbauer, Jirovetz, Jager, Plank & Dietrich, 1993).
Their physiological mechanisms are protean particularly in the CNS, attributable to their lipophilicity, and include effects on ion channels, neurotransmitter, odorant, and tastant receptors, among others (Buchbauer, 2010). Terpenoids, particularly monoterpenoids, are highly bioavailable via inhalation (Falk, Hagberg, Lof, Wigaeus-Hjelm, & Wang, 1990; Falk, Lof, Hagberg, Hjelm, & Wang, 1991; Falk-Filipsson, Lof, Hagberg, Hjelm, & Wang, 1993). Terpenoid concentrations in cannabis flowers were previously commonly reported in the 1% range, but up to 10% within trichomes (Potter, 2009), but this situation has changed due to selective breeding, such that flower concentrations of 3.5% (Fischedick, Hazekamp, et al., 2010) or even higher in modern chemovars are now encountered.
Many have argued that cannabis is primarily a botanical delivery device for THC, while others have espoused amore holistic assessment (seeMcPartland, Guy, & Di Marzo, 2014; Russo, 2011 for a broader discussion). Certainly, medical consumersmust fall into the latter group, as sales figures for herbal cannabis overwhelm those for THC (Marinol®) as a pure compound. Sativex®, a standardized oromucosal whole cannabis extract that is now approved as a prescription in 29 countries, was purposely designed to incorporate terpenoids, which comprise 6%–7% of total cannabinoid (Guy & Stott, 2005).
While controlled double-blind trials exploring cannabinoid–terpenoid interactions have yet to take place and are sorely required, observational information has been offered: limonene added to THC enhanced the experience to be more “cerebral and euphoric,” while myrcene rendered THC more “physical, mellow, sleepy.” The three together were considered more “cannabimimetic” than THC alone (Name Withheld, 2006), THC taken in isolation being more dysphoric than euphoric (Calhoun et al., 1998), and displaying a much narrower therapeutic index than whole cannabis (Russo, 2011; Sellers et al., 2013). Clinical trial data comparing rates of adverse events also favor cannabis extracts over THC (Russo, 2013).
While the following will summarize prior publications, emphasis will be placed on newer findings and agents not previously examined in relation to cannabis pharmacology. Unless otherwise indicated, all the agents are Generally Recognized As Safe (GRAS) by the US Food and Drug Administration (FDA) and/or are approved as food additives by the Flavor and Extract Manufacturers’ Association (FEMA). According to a recent publication (Giese, Lewis, Giese, & Smith, 2015), 50 cannabis terpenes are routinely encountered in North American chemovars, but 17 are most common, all of which are discussed herein. Of these, several predominate to form eight “Terpene Super Classes”: myrcene, terpinolene, ocimene, limonene, α-pinene, humulene, linalool, and β-caryophyllene (BCP). Similarly, Fischedick (2017) analyzed cannabis samples from a single California cannabis dispensary over the course of a year, and identified five terpenoid groups based on predominant content: myrcene, terpinolene, myrcene/limonene, caryophyllene, and bisabolol.
4. CANNABIS MONOTERPENOIDS
4.1 β-Myrcene
β-Myrcene is the most prevalent terpene in modern cannabis chemovars in the United States (Giese et al., 2015) and in Europe (Hazekamp, Tejkalova, & Papadimitriou, 2016), and is likely most responsible for sedative effects of many of the common preparations in commerce.
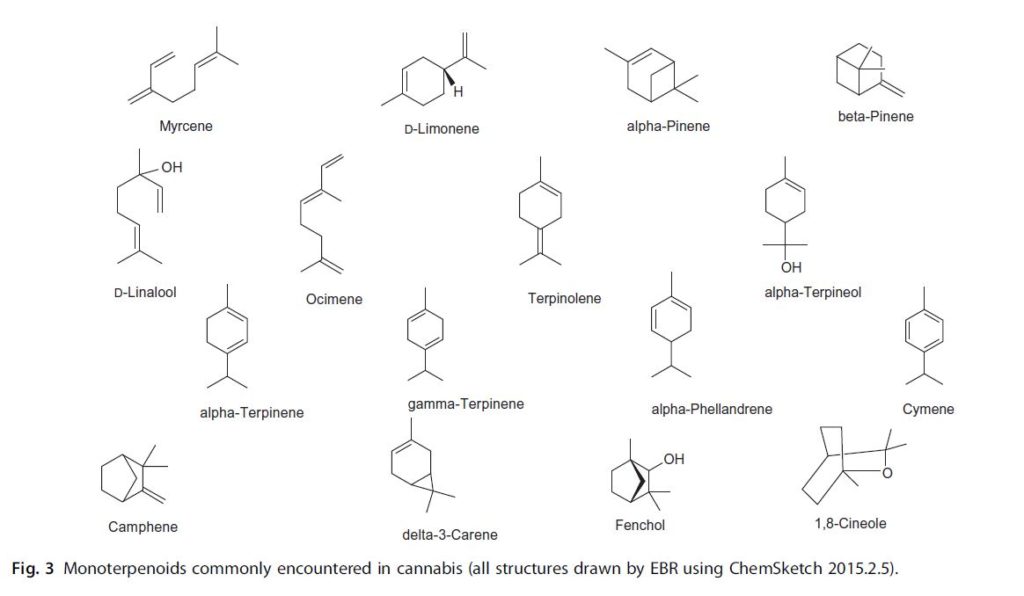
Fig. 3 Monoterpenoids commonly encountered in cannabis (all structures drawn by EBR using ChemSketch 2015.2.5).
As previously reviewed (Russo, 2011), myrcene is anti-inflammatory via prostaglandin E-2 (PGE-2) (Lorenzetti, Souza, Sarti, Santos Filho, & Ferreira, 1991), blocks carcinogenic effects of aflatoxin in the liver (De-Oliveira, Ribeiro-Pinto, & Paumgartten, 1997), and is analgesic in mice, an activity that is abrogated by naloxone, the μ-opioid inverse agonist, suggesting a narcotic effect mediated by α-2 adrenoreceptors (Rao, Menezes, & Viana, 1990). This is less surprising recognizing that myrcene is one sedative agent of hops (Humulus lupulus) (Bisset & Wichtl, 2004). Additionally, it produces muscle relaxant effects in mice, and prolonged barbiturate sleep time (do Vale, Furtado, Santos, & Viana, 2002). These findings seem to explain the phenomenology of “couch-lock” commonly attributed to modern cannabis chemovars by its consumers.
More recent studies expand on these findings. In mice (Paula-Freire, Andersen, Gama, Molska, & Carlini, 2014), myrcene 10 mg/kg po (equivalent to 0.81 mg/kg in humans) (Reagan-Shaw, Nihal, &Ahmad, 2008) significantly increased paw lick latency in the hot-plate test, and reduced pain behavior in both phases of the formalin test. Interestingly, the duration of analgesic effect exceeds that of morphine (4 h) and once again, was abrogated by naloxone administration, supporting an opioid-related mechanism of action.
In human chondrocyte culture, myrcene inhibited NO production by IL-1β with an IC50 of 37.3 μg/mL, and at 50 μg/mL, lowered IL-1β- induced iNOS mRNA and protein by 78% (Rufino et al., 2015), suggesting therapeutic application in osteoarthritis.
In rats (Bonamin et al., 2014), oral myrcene 7.5 mg/kg (equivalent to 1.2 mg/kg in humans) demonstrated notable effects against peptic ulcers: decreased lesions in stomach and duodenum, increased mucus production, and mucosal malondialdehyde levels indicative of oxidative damage, decreased superoxide dismutase, but increased glutathione peroxidase, glutathione reductase, and total glutathione in the tissues. Similarly, in mice, myrcene 200 mg/kg ip (equivalent to 16.2 mg/kg in humans) for 10 days prevented ischemic/reperfusion oxidative injury via increases in glutathione, glutathione peroxidase, and superoxide dismutase, decreasing thiobarbituric acid reactive substances, and eliminating cerebral apoptosis and other histological changes (Ciftci, Oztanir, & Cetin, 2014). This activity suggests the possibility of synergistic benefits with the neuroprotective antioxidant effects of THC and CBD (Hampson et al., 1998; Lafuente et al., 2011).
4.2 D-Limonene
Limonene is a cyclic monoterpene common to citrus rinds and is common in nature, though more sporadically encountered in contemporary cannabis. It displays high bioavailability with 70% absorption after human pulmonary administration (Falk-Filipsson et al., 1993), is rapidly metabolized (Falk-
Filipsson et al., 1993), but accumulates in adipose tissues and the brain, with an estimated human lethal dose of 0.5–5 g/kg. It is nonsensitizing (Von Burg, 1995).
Limonene is the parent compound to the entire family of monoterpenoids in the plant, and its biosynthetic enzyme, limonene synthase and others in cannabis are promiscuous in their substrates with various terpenoid end-products (Booth et al., 2017), via regulatory mechanisms that remain to be elucidated. Experiments in mice confirm limonene to be strongly anxiolytic, boosting serotonin levels in prefrontal cortex, and dopamine in hippocampus mediated via 5-HT1A receptors (Carvalho-Freitas & Costa, 2002; Komiya, Takeuchi, & Harada, 2006; Pultrini Ade, Galindo, & Costa, 2006). Orange terpenes, primarily limonene, boosted mouse motility after inhalation by 35.25%, while decreasing activity after caffeine 33.19% (Buchbauer et al., 1993). Human clinical work supports these activities, as a study in Japan (Komori, Fujiwara, Tanida, Nomura, & Yokoyama, 1995), demonstrated that depressed patients exposed to citrus scent experienced normalization of Hamilton Depression Scores (HADS), allowing discontinuation of antidepressants in 9/12 hospitalized patients. Additionally, immune stimulation (CD4/8 ratio normalization) was documented. Limonene has an impressively supportive history as an “antidote” to excessive psychoactive adverse events produced by THC (Russo, 2011).
Limonene demonstrated prominent antibiotic effects vs S. aureus and Pseudomonas aeruginosa (Onawunmi, Yisak, & Ogunlana, 1984). Recently, concentrations of 400 μg/mL inhibited biofilm formation of the pathogen Streptococcus pyogenes SF370 and S. nutans, which produces dental caries, downregulating various genes mediating surface-associated proteins (Subramenium, Vijayakumar, & Pandian, 2015). Considering that cannabinoids also interfere with quorum-sensing in biofilm formation (Soni, Smoum, Breuer, Mechoulam, & Steinberg, 2015), cannabinoid/terpenoid synergy in this mechanism of action is certainly likely.
Citrus EOs were an effective treatment against dermatophytes (Sanguinetti et al., 2007; Singh et al., 2010), and display radical scavenging abilities (Choi, Song, Ukeda, & Sawamura, 2000). Two citrus Eos also suppressed Propionibacterium acnes, the pathogen in acne (MIC 0.31 μL/mL), more powerfully than triclosan (Kim et al., 2008), while simultaneously lowering TNF-α production.
Limonene also demonstrates chemotherapeutic properties, inducing apoptosis of breast cancer cells among others. It was utilized in high doses in Phase II RCTs (Vigushin et al., 1998), with good safety, but less impressive efficacy. A more recent study in humans demonstrated that, in women with preoperative breast cancer, an oral intake of 2 g of D-limonene a day produced a mean concentration of 41.3 μg/g of biopsy breast reduced cyclin D1 expression that could lead to cell-cycle arrest and decreased proliferation (Miller et al., 2013).
A blood orange (Citrus sinensis) volatile emulsion that was 95.35% D-limonene at 100 ppm induced apoptosis in Bcl-2 human colon cancer cells, activating p38 and inhibiting Akt, and inhibited the angiogenesismarker, vascular endothelial growth factor 80%, decreased cell migration, downregulated
MMP-9 expression, and reduced tube formation (Chidambara Murthy, Jayaprakasha, & Patil, 2012). Limonene’s primarymetabolite, perillic acid, also has cytotoxic effects, and additionally produces antianxiety effects in rat brain (Fukumoto et al., 2008).
A patent has been filed based on the ability of limonene to ameliorate gastro-esophageal reflux (Harris, 2010) and a commercial capsule preparation is available. Limonene 10 mg/kg po reduced hyperalgesia in mice induced by intrathecal administration of HIV glycoprotein toxin gp120, as well as prevented increases in IL-1β and IL-10 levels (Piccinelli et al., 2017). Mechanical sensitivity induced by TNF-α, was prevented, as was IL-1β cold sensitivity.
Limonene 10 mg/kg po reduced inflammation scores, weight loss, and TNF-α in ibuprofen-induced rat colitis, as well as decreased peripheral IL-6 inflammatory marker in elderly humans receiving a daily supplement that was 95% limonene for 56 days (d’Alessio et al., 2013).
At high concentrations, limonene prevented oxidative damage in human lens epithelial cells via regulation of caspase-3 and -9, Bax, and Bcl-2, as well as inhibition of p38 MAPK phosphorylation (Bai, Zheng, Wang, & Liu, 2016), suggesting therapeutic use to prevent cataracts.
Limonene is an agonist at A2A adenosine receptors (Park, Lee, Yaoyao, Jun, & Lee, 2011) and could synergize activity with both THC (direct activator) and CBD (uptake inhibitor via competition for the nucleotide binding site of the ENT1 transporter) (Carrier et al., 2006), a relationship that is now the subject of active research.
Limonene 50 μM increased mitochondrial biogenesis, activated the AMPK energy regulator, increased brown adipocyte markers PGC-1α UCP1, and induced “browning” of 3T3-L1 adipocytes by activating β-3-AR and ERK signaling pathway (Lone & Yun, 2016), suggesting a putative role in obesity treatment. Certainly, interesting synergies are possible with the anorexic effects of CBD and THCV (McPartland et al., 2015), and modulatory effects of THC on weight and microbiome balance (Cluny, Keenan, Reimer, Le Foll, & Sharkey, 2015).
4.3 β-Ocimene
Ocimene is one of the most common monoterpenes found in nature. In the field of botanical medicine, there is an association of β-ocimene in EOs with anticonvulsant activity, antifungal activity, antitumor activity, and pest resistance (Bomfim et al., 2016; Cascone et al., 2015; Sayyah, Nadjafnia, & Kamalinejad, 2004). Ocimene is also a volatile pheromone important for the social regulation of honeybee colonies. The commercial applications of exploiting that attraction to produce “cannabis honey” have not been missed by the cannabis industry emerging in the United States, and subsequently by law enforcement agencies to detect illicit drugs by “trained honeybees,” which were proposed to replace sniffer dogs in 2015 (Kennell, 2016; Maisonnasse, Lenoir, Beslay, Crauser, & Le Conte, 2010; Schott, Klein, & Vilcinskas, 2015).
Significant ocimene content is being reported by medical cannabis laboratories in California and Washington State (Elzinga et al., 2015). Ocimene is also a major component of the EO of cannabis varieties developed by the international medical cannabis producer, Bedrocan, which supplies standardized cannabis to pharmacies in Europe (Fischedick, Van Der Kooy, & Verpoorte, 2010). The effects and associations of cannabinoid and ocimene co-administration remain unclear but warrant further attention.
4.4 γ-Terpinene
This cyclic monoterpene is common to Eucalyptus spp., and to EO of cumin (Cuminum cyminum, 32%), whereas it is a minor component in cannabis. In mice, oral pretreatment with of 25–50 mg/kg (equivalent to 2–4 mg/kg human) inhibited extravasation of fluid in an acetic acid microvascular permeability model, reduced peritonitis after carageenan, neutrophil migration, and production of interleukin-1β and TNF-α vs controls, as well as lung inflammation after acute injury, thus demonstrating broad anti-inflammatory effects (Ramalho, Pacheco de Oliveira, Lima, Bezerra-Santos, & Piuvezam, 2015). γ-Terpinene demonstrated little antioxidant or antiproliferative activity in a recent experiment (Fitsiou et al., 2016).
4.5 α-Terpinene
A major component of tea tree oil (Melaleuca alternifolia, 13%) it is found in low concentrations in cannabis. It inhibited oxidation of LDL and linoleic acid and was potent as a scavenger of DPPH radicals (Tisserand & Young, 2014). It demonstrated modest activity as a synergist to diminazene aceturate in treatment of Trypanasoma evansi, a protozoal pathogen of horses and other animals (Baldissera et al., 2016).
4.6 α-Terpineol
Terpineol is a cyclic monoterpenoid alcohol (Bhatia, Letizia, & Api, 2008).
Its inhalation diminished mouse motility 45% (Buchbauer et al., 1993). It displayed dose-dependent antibiotic efficacy vs S. aureus, S. epidermidis, and P. acnes (Raman, Weir, & Bloomfield, 1995), among others, particularly in its customary vehicle of tea tree oil (M. alternifolia) (Carson & Riley, 1995). An MIC of 0.78 μL/mL was noted on Escherichia coli, with observed cell wall and membrane rupture (Li et al., 2014). α-Terpineol 100 μg/disk produced significant zones of inhibition in culture of four drug-resistant Helicobacter pylori cultures (Miyamoto, Okimoto, & Kuwano, 2014). Moderate effects against two strains of Plasmodium falciparum malaria were noted in an EO with major terpineol component (Campbell, Gammon, Smith, Abrahams, & Purves, 1997).
The small cell lung cancer cell line NCI-H69 was sensitive to α-terpineol at a high dose (IC50 approximately 260 μM) via suppression of NF-κB signaling (Hassan, Gali-Muhtasib, Goransson, &Larsson, 2010). In a U937 leukemia cell line, α-terpineol reduced LPS-induced cytokine production of IL-1β, IL-6, and IL-10, but not TNF-α (Nogueira, Aquino, Rossa Junior, & Spolidorio, 2014).
Nociceptive behavior in mice was significantly reduced by doses of 25 mg/kg ip and above on early and late paw licking post formalin, writhing after ip acetic acid, and after paw injections of glutamate or capsaicin, without motor impairment (Quintans-Junior et al., 2011). Similarly, 50–100 mg/kg ip dosing in mice inhibited hyperalgesia postcarageenan or TNF-α, PGE2, or DA administration, and neutrophil migration in a pleurisy model (de Oliveira et al., 2012).
It was reported that fatty liver was produced in mice after daily injections of 10 or 500 mg/kg ip of α-terpineol for 2 weeks (Choi, Sim, Choi, Lee, & Lee, 2013), an exposure level likely never attainable with a cannabis-based medicine. Two recent studies from Iran are of interest. Pretreatment with α-terpineol 5–20 mg/kg ip significantly reduced jumping behavior typical of withdrawal effect in mice rendered morphine-dependent (Parvardeh, Moghimi, Eslami, & Masoudi, 2016), while 20–40 mg/kg ip doses reduced the development of tolerance to morphine analgesia. These results suggest possible synergy of this ingredient with other cannabis components attenuating addiction: CBD and BCP (Russo, 2011). Higher doses of α-terpineol (50–200 mg/kg ip) in rats subjected to cerebral ischemia improved spatial learning in a water maze vs controls, restored hippocampal long-term potentiation, and lowered malondialdehyde levels indicative of lipid peroxidation (Moghimi, Parvardeh, Zanjani, & Ghafghazi, 2016). This activity certainly suggests the possibility of synergistic benefit in conjunction with benefits ascribed to CBD in similar experiments in newborn pigs (Lafuente et al., 2011).
4.7 α-Pinene
α-Pinene, a bicyclic monoterpene, is the most widely distributed terpenoid in Nature (Noma & Asakawa, 2010), but this versatile therapeutic agent is unfortunately represented in lower concentration in modern cannabis chemovars, although it is reportedly relatively abundant in the “Blue Dream” chemovar in Southern California (Backes, 2014). It has high bioavailability via inhalation (60%) with rapid metabolism and redistribution (Falk et al., 1990).
Its pharmacological effects are legion: anti-inflammatory via PGE-1 (Gil,Jimenez, Ocete, Zarzuelo, & Cabo, 1989), bronchodilator in humans at low exposure levels (Falk et al., 1990), antibiotic in EO that was equally effective as vancomycin against MRSA and other resistant bacteria (Kose, Deniz, Sarikurkcu, Aktas, & Yavuz, 2010) (MIC 125 μg/mL) in an EO of Salvia rosifolia composed of 34.8% pinene, and was the most potent compound in a tea tree EOvs P. acnes and Staph spp. (Raman et al., 1995). Efficacy was also noted for α-pinene for MRSA, Cryptococcus neoformans and Candida albicans biofilms (Rivas da Silva et al., 2012). α-Pinene dramatically increased antibiotic efficacy by lowering theMIC of ciprofloxacin, erythromycin, and triclosan against the gastroenteritis pathogen, Campylobacter jejuni, by promoting cmeABC and Cj1687 antimicrobial efflux genes, decreasing bacterial membrane integrity, and disrupting heat-shock responses (Kovac et al., 2015).
It was also beneficial against Leishmania amazonensis promastigotes (IC50 19.7 μg/mL) and axenic and intracellular amastigote forms (IC50 43.9 and 38.1 μg/mL) (Rodrigues et al., 2015). α-Pinene demonstrated larvicidal activity against Anopholes subpictus, vector of malaria (LC50 [lethal concentration] 32.09 μg/mL), Aedes albopictus, vector of dengue (LC50 34.09 μg/mL), and Culex tritaeniorhynchus, vector of Japanese encephalitis (LC50 36.75 μg/mL) (Govindarajan, Rajeswary, Hoti, Bhattacharyya, & Benelli, 2016).
Pinene increased mouse motility after inhalation 13.77% (Buchbauer et al., 1993). Its greatest therapeutic value may derive from its acetylcholinesterase inhibition (Perry, Houghton, Theobald, Jenner, & Perry, 2000), producing an IC50 of 0.44 mM (Miyazawa & Yamafuji, 2005), which serves to reduce or eliminate one of the primary adverse events associated with THC, that of short-term memory impairment. This ability may also serve admirably in treatment of dementia, a syndrome in which THC has already produced benefits in counteracting agitation (Russo, Guy, & Robson, 2007; Volicer, Stelly, Morris, McLaughlin, & Volicer, 1997).
Inhalation of α-pinene in mice at 10 μL/L concentration produced an anxiolytic effect in the elevated plus maze, with general brain distribution and increase in tyrosine hydroxylase mRNA in the midbrain (Kasuya et al., 2015). In chronic inhalation over 5 days, anxiolytic effects were maintained (Satou, Kasuya, Maeda, & Koike, 2014). α-Pinene has also been suggested as a modulator of THC overdose events (Russo, 2011), with historical anecdotes supporting its use as an antidote to cannabis intoxication. α-Pinene at a concentration of 2 μg/mL produced 69% protection in rat astrocytes against H2O2-induced cell death (Elmann et al., 2009).
Chronic pinene exposure led to decreased melanoma growth in mice at 180 ng/L (1 ppm) in ambient air, a dose too low to directly affect tumor (Kusuhara et al., 2012). This mental health-promoting effect attributed here to pinene exposure, is known in Japan as “Shinrin-yoku” or “forest bathing.” In contrast, a direct synergistic and isobolographic benefit was observed with α-pinene in combination with paclitaxel vs nonsmall-cell A549 lung carcinoma cells with evidence of apoptosis (Zhang et al., 2015). Α Pinene inhibited BEL-7402 human hepatoma cell growth 79.3%, both time and dose dependently over 3 days at 8 mg/L concentration (Chen et al., 2015), causing cycle arrest in G2/M phase, a decrease in tumor xenografts vs control (P<0.01), and equivalent to that from 5-flurouracil, an increase in Chk1 and -2 expression, indicative of DNA damage leading to cell death.
4.8 β-Pinene
A bicyclic monoterpene isomer, β-pinene is commonly encountered in conjunction with α-pinene. It proved to have equal antibiotic efficacy to α-pinene against S. aureus (MRSA), and C. neoformans and C. albicans biofilms (Rivas da Silva et al., 2012). Like its isomer, β-pinene demonstrated the ability to synergize with paclitaxel vs nonsmall-cell A549 lung carcinoma cells with evidence of apoptosis (Zhang et al., 2015), but unlike α-pinene, it failed to prevent astrocyte damage by H2O2 (Elmann et al., 2009). Little additional pharmacological research has been evident otherwise on the pure compound, particularly with regard to its psychopharmacology.
4.9 Linalool
Linalool is a noncyclic monoterpenoid that is commonly extracted from lavender (Lavandula spp.), rose (Rosa spp.), basil (Ocimum basilicum), and neroli oil (Citrus aurantium). The psychotropic anxiolytic activity has been reviewed in detail (Russo, 2001, 2011). Linalool has established sedative, antidepressant, anxiolytic, and immune potentiating effects and can represent a significant portion (<6%) of the EO of cannabis (McPartland & Russo, 2001). This terpene can also have analgesic and anticonvulsant effects (Batista et al., 2010; Leal-Cardoso et al., 2010; Peana et al., 2006; Russo, 2011). It is also antinociceptive at high doses in mice via ionotropic glutamate receptors (Batista et al., 2008). Linalool demonstrated anticonvulsant and antiglutamatergic activity, and reduced seizures as part of O. basilicum
EO after exposure to pentylenetetrazole, picrotoxin, and strychnine (Elisabetsky, Marschner, & Souza, 1995; Ismail, 2006). Furthermore, linalool decreased K+-stimulated glutamate release and uptake in mouse synaptosomes (Silva Brum, Emanuelli, Souza, & Elisabetsky, 2001). Recent reports support the possibility that small concentrations found in certain cannabis chemovars may exert anticonvulsant benefits in human patients (Russo, 2016b; Sulak, Saneto, & Goldstein, 2017).
Linalool alone demonstrated an MIC of 0.625 μL/mL on P. acnes (Kim et al., 2008). Linalool in ambient air decreased mouse motility 73%, confirming its potent sedative effects (Buchbauer et al., 1993). In traditional aromatherapy, linalool is the likely suspect in the remarkable therapeutic capabilities of lavender EO to alleviate skin burns without scarring (Gattefosse, 1993). Pertinent to this, the local anesthetic effects of linalool are equal to those of procaine and menthol (Ghelardini, Galeotti, Salvatore, & Mazzanti, 1999; Re et al., 2000). Another explanation would be its ability to produce hot-plate analgesia in mice (P<0.001) that was reduced by administration of an adenosine A2A antagonist (Peana et al., 2006). This terpene can also influence CYP enzymes in rat liver, suggesting that it can alter the pharmacokinetics of cannabis administration (Noskova, Dovrtelova, Zendulka, Rˇ emı´nek, & Jurica, 2016). Linalool displays powerful antileishmanial activity, and as a presumed lavender EO component, decreased morphine opioid usage after inhalation vs placebo (P¼0.04) in gastric banding in morbidly obese surgical patients (do Socorro et al., 2003; Kim et al., 2007). Linalool incorporated nanoparticles are being explored as a novel anticancer agent (Han et al., 2016).
4.10 Camphene
Camphene is a cyclic monoterpene common to conifers, especially Douglas fir (Pseudotsuga menziesii), and is present in many cannabis chemovars in low titer. In an ointment with menthol and other EOs, camphene reduced experimentally induced bronchospasm in animals, suggesting application in human chronic obstructive pulmonary disease (Schafer & Schafer, 1981). Camphene administered to hyperlipidemic rats at 30 μg/g (equivalent to 4.87 mg/kg in humans) led to a 54.5% decrease in total cholesterol, 54% in LDL-cholesterol, and 34.5% in triglycerides (all P<0.001) (Vallianou, Peroulis, Pantazis, & Hadzopoulou-Cladaras, 2011). Reductions in cholesterol in HepG2 cells paralleled those attained with mevinolin, but in contrast, camphene seemingly worked independently of HMG-CoA reductase inhibition. Synergy of camphene with other components of Chios mastic gum (Pistacia lentiscus) was also observed. In subsequent work (Vallianou & Hadzopoulou-Cladaras, 2016), camphene inhibited cholesterol production 39% at 100 μM in HepG2 cells, while also decreasing triglycerides 34% and increasing apolipoprotein AI expression, likely mediated via SREBP-1 upregulation and MTP inhibition. Camphene displayed weak antinociceptive effects on acetic acidinduced writhing in mice at 200 mg/kg (Quintans-Junior et al., 2013), but prevented AAPH-induced lipoperoxidation at 0.01 μg/mL, and demonstrated antioxidant activity and superoxide radical inhibition at the same concentration.
Camphene supplemented to the high-fat diet of mice at the high dose of 200 mg/kg/day (corresponding to 16 mg/kg/day in humans) reduced 17% body weight reduction vs controls (Kim, Choi, Choi, Choi, & Park, 2014), and increased adiponectin levels and receptor mRNA expression in liver.
Camphene induced apoptosis in a variety of cancer cell lines, notably B16F10-Nex2 melanoma with an IC50 of 71.2 μg/mL (Girola et al., 2015) and produced chromatin condensation, shrinkage of cells, apoptotic body formation, and fragmentation of nucleus and activation of caspase-3. It was also active against grafted tumor with peritumoral injection (10 mg/mL) in mice.
Camphene was utilized as a porogen for the production of nano/ macroporous polycaprolactone microspheres for injectable cell delivery (Kim, Hwang, & Shin, 2016).
4.11 Terpinolene
Terpinolene is a cyclic monoterpene, common to Pinus spp., but richest in parsnip EO (Pastinaca sativa 69%) (Tisserand&Young, 2014). It is a common component of some commercial cannabis chemovars (Giese et al., 2015), its presence is said to be characteristic of “sativa” types (Hazekamp et al., 2016). Terpinolene has been demonstrated to prevent LDL oxidation, of interest in treatment of atherogenesis and coronary artery disease (Grassmann, Hippeli, Spitzenberger, & Elstner, 2005).
It was sedative in mice at 0.1 mg, reducing motor activity to 67.8% (Ito & Ito, 2013), whereas subjective reports in humans suggest greater stimulation in terpinolene-rich cannabis chemovars (data on file, Napro Research 2016), possibly attributable to cholinesterase inhibition effects in the presence of THC, a pharmacological effect measured with IC50 at 156.4 μg/mL (Bonesi et al. , 2010). At a concentration of 0.05%, terpinolene markedly reduced AKT1 expression in K562 human CML cells and significantly stimulated apoptosis (Okumura, Yoshida, Nishimura, Kitagishi, & Matsuda, 2012). At extreme dosing (>50 mg/L), terpinolene demonstrated marginally greater antiproliferative effects against neuroblastoma as compared to neuronal cell lines (Aydin, Turkez, & Tasdemir, 2013). Over a similar dosage range, it showed antioxidant effects in human lymphocytes (Turkez, Aydin, Geyikoglu, & Cetin, 2015). Terpinolene is reportedly also antifungal and larvicidal (Aydin et al., 2013). A subactive antinociceptive and antiinflammatory dosage of 3.125 mg/kg po in rats synergized with diclofenac, and reduced hyperalgesia, an effect blocked by ketanserin, suggesting mediation via 5-HT2A receptors (Macedo et al., 2016).
4.12 α-Phellandrene
A cyclic monoterpene, α-phellandrene is widespread in nature, but rich in frankincense (Boswellia sacra), comprising 42% of the EO (Tisserand & Young, 2014). It produced cholinesterase inhibition with an IC50 of 120.2 μg/mL (Bonesi et al., 2010). Multiple assays in mice (Lima et al., 2012) demonstrated antinociceptive effects: acetic acid-induced abdominal writhing (3.125 mg/kg/po or 0.25 mg/kg human equivalent), both phases of the formalin test (50 mg/kg/po, or 0.54 mg/kg human), capsaicin injection (3.125 mg/kg/po, or 0.25 mg/kg human equivalent), glutamate injection (12.5 mg/kg/po, or 1 mg/kg human) and carageenan injection (only at 3 h at 25 mg/kg/po, or 2 mg/kg human). Effects were blocked by multiple agents, suggesting mediation by glutamatergic, opioid, nitrergic, cholinergic, and adrenergic mechanisms. In rats, phellandrene at 10 mg/kg/d po (1.6 mg/kg human equivalent) prevented spared nerve injury-induced mechanical and cold hyperalgesia, while also demonstrating an antidepressant effect in reducing immobility in the forced swim test 85%, but without decreasing locomotor activity in the open field (Piccinelli et al., 2015).
While not demonstrating antimicrobial effects per se, phellandrene mildly stimulated macrophage proliferation in mice via Mac-3 and promoted function in vivo (Lin et al., 2013), suggesting ability to suppress intracellular bacterial growth. Subsequent work demonstrated a wide variety of effects on gene expression affecting DNA repair, cell cycle, and apoptosis in WEHI-3 murine leukemia cells (Lin et al., 2015, 2014).
At 30 μM concentration with 24 h of exposure, 15.8% of human liver tumor J5 cells became necrotic, possibly due to depletion of ATP (Hsieh et al., 2014). Subsequently, findings were attributed to multiple pathways: regulation of mTOR, LC-3II expression, p53 signaling and NF-κB activation (Hsieh et al., 2015). In carageenan injections in rodents, phellandrene 50 mg/kg po pretreatment induced neutrophil migration inhibition, and TNF-α release (both P<0.001) (Siqueira et al., 2016) and decreased mast cell degranulation (P<0.05), suggesting possible applications in arthritic and allergic conditions.
4.13 γ-Cadinene
A bicyclic sesquiterpene, while more common in other EOs, it is found at low concentration in current cannabis chemovars tested (Hazekamp et al., 2016). Cadinene demonstrated larvicidal activity against Anopholes stephensi, vector of malaria (LC50 [lethal concentration] 8.23 μg/mL), Aedes aegypti, vector of dengue (LC50 9.03 μg/mL), and Culex quinquefaciatus, vector of filariasis (LC50 9.86 μg/mL) (Govindarajan, Rajeswary, & Benelli, 2016). Little additional pharmacological data is available on the isolated compound.
4.14 Δ3-Carene
A bicyclic monoterpenoid alkene most associated with turpentine from conifers, it is also prevalent in white pepper (Piper nigrum, 25%) (Tisserand & Young, 2014), but is found in low concentration in cannabis.
Studies from Scandinavia in sawmills have documented high-exposure human irritancy reactions in skin and lungs, with pulmonary intake and slight increased airway resistance at 450 mg/m3 exposure (Falk et al., 1991), with rapid metabolism and high adipose tissue affinity. The occupational exposure limit recommendation in Sweden for it or other turpentine components is 150 mg/m3 (Kasanen et al., 1999). Carene hydroperoxide was noted to be an allergen (Edman et al., 2003), and skin sensitization in guinea pigs at very high concentrations increased airway reactivity (Lastbom, Boman, Johnsson, Camner, & Ryrfeldt, 2003). Carene concentrations, along with limonene and pinene, are common volatile organic compounds elevated in new home construction (Krol, Namiesnik, & Zabiegala, 2014). Carene was rapidly absorbed, distributed, and metabolized in human volunteers after oral administration (Schmidt, Belov, & Goen, 2015). A low concentration (5 μM) stimulated mineralization in mouse osteoblastic cells by increasing protein expression; activation of MAP kinases; and expression of osteoblast genes, osteopontin, and type I collagen (Jeong, Kim, Min, & Kim, 2008), suggesting a possible therapeutic role in osteoporosis treatment. Carene demonstrated larvicidal activity against Anopholes stephensi, vector of malaria (LC50 [lethal concentration] 16.37 μg/mL), A. aegypti, vector of dengue (LC50 17.91 μg/mL), and C. quinquefaciatus, vector of filariasis (LC50 19.5 μg/mL) (Govindarajan, Rajeswary, Hoti, et al., 2016; Govindarajan, Rajeswary, et al., 2016). Carene content was judged to be a marker of “sativa” cannabis chemovars (Hazekamp et al., 2016).
4.15 ρ-Cymene
A cyclic monoterpene, common to thyme (Thymus vulgaris) (27.4%), but a minor component in cannabis, ρ-cymene was active against Bacteroides fragilis, C. albicans, and Clostridium perfringens (Carson & Riley, 1995). It was sedative in mice at 0.04 mg in air, reducing motor activity to 47.3% of baseline (Ito&Ito, 2013). Additionally, it statistically significantly reduced acetic acid-induced writhing and both phases of formalin-induced pain in mice at 50 mg/kg (Quintans-Junior et al., 2013). It showed little antioxidant or antiproliferative effects in a recent study (Fitsiou et al., 2016).
4.16 Fenchol
A bicyclic monoterpenoid, fenchol (or fenchyl alcohol) is an FDA-approved flavor additive and rated GRAS by FEMA (Bhatia, McGinty, Letizia, & Api, 2008). Oral doses above 2 g/kg were fatal in rats, demonstrating lethargy, ataxia, flaccidity, and coma, whereas a 4% cutaneous application in humans was nonsensitizing. It is common to basil (O. basilicum), and to California cannabis chemovars (Giese et al., 2015), but in low concentrations, such that the noted toxicity would be unlikely a factor even in concentrates.
4.17 1, 8-Cineole (Eucalyptol)
This bicyclic monoterpenoid ether is a major component of Eucalyptus spp. EOs, and is largely responsible for their pharmacology (Barbosa, Filomeno, & Teixeira, 2016). A prior review (McPartland & Russo, 2001) noted its myriad activities including increasing cerebral blood flow after inhalation, increasing rat locomotion, and as an antiinflammatory, analgesic, antibiotic, antifungal, and antiviral against Herpes simplex 2, but it is barely present in modern cannabis chemovars (Hazekamp et al., 2016).
5. CANNABIS SESQUITERPENOIDS
5.1 β-Caryophyllene
BCP, a bicyclic sesquiterpenes alkene, is the most common terpenoid in cannabis extracts, and is nearly ubiquitous in food in the food supply. The extensive potent and various pharmacological activities for BCP summarized below, are rarely noted for any individual compound that also has a wide therapeutic index, safety, and low toxicity. BCP acts as a selective full agonist at CB2 with strong potency (100 nM), and its anti-inflammatory effects are reduced in CB2 knockout mice (Gertsch, 2008). BCP activity at CB2 has been confirmed in rodent models of nociception and pain (Katsuyama et al., 2013; Paula-Freire et al., 2014), colitis (Bento et al., 2011), and nephrotoxicity (Horva´th, Mukhopadhyay, Hasko, & Pacher, 2012). Russo (2011) proposes mechanisms whereby BCP synergizes with THC to impart antipruritic effects and gastric cytoprotection, and with CBD to impart antiinflammatory benefits. CB2 agonists (likely including caryophyllene) have been shown to reduce drug administration (cocaine) and improve scores of depression and anxiety in animal models (Bahi et al., 2014; Onaivi et al., 2008; Xi et al., 2011). BCP demonstrated larvicidal activity against A. subpictus, vector of malaria (LC50 [lethal concentration] 41.66 μg/mL), A. albopictus, vector of dengue (LC50 44.77 μg/mL), and C. tritaeniorhynchus, vector of Japanese encephalitis (LC50 48.17 μg/mL) (Govindarajan, Rajeswary, Hoti, et al., 2016; Govindarajan, Rajeswary, et al., 2016). As a monotherapeutic agent, BCP providesmany other benefits, reviewed by Fidyt, Fiedorowicz, and Strza˛dała (2016).According to an exhaustive review, BCP activates peroxisome proliferated activator receptors (PPARs) isoforms, inhibits pathways triggered by the activation of toll-like receptor complexes (i.e., CD14/TLR4/MD2), reduces
immunoinflammatory processes, and exhibits synergy with μ-opioid receptor pathways (Sharma et al., 2016). Additionally, BCP is a potent antagonist of homomeric nicotinic acetylcholine receptors (7-nAChRs) and devoid of effects mediated by serotonergic and GABAergic receptors. BCP modulates numerous molecular targets by altering their gene expression, signaling pathways, or through direct interaction. Basic experiments have demonstrated strong evidence for cardioprotective, hepatoprotective, gastroprotective, neuroprotective, nephroprotective, antioxidant, antiinflammatory, antimicrobial, and immunemodulator activities. Thus, it has shown potent therapeutic promise in neuropathic pain, neurodegenerative, and metabolic diseases. A recent publication extends its therapeutic potential to protection fromalcoholic steatohepatitis via antiinflammatory effects and alleviation of metabolic disturbances (Varga et al., 2017).
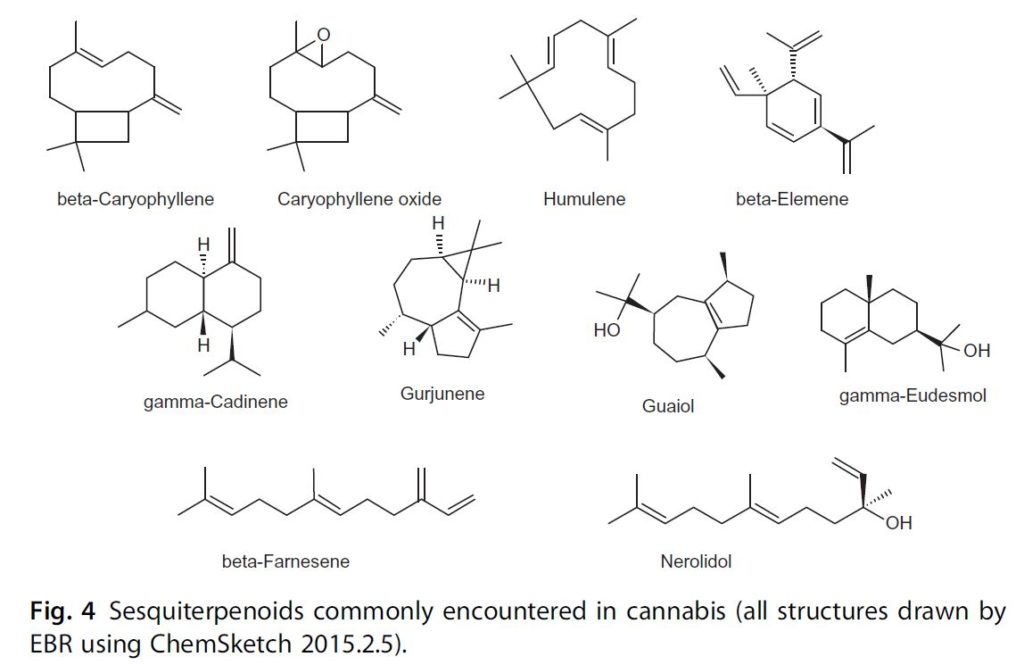
Fig. 4 Sesquiterpenoids commonly encountered in cannabis (all structures drawn by EBR using ChemSketch 2015.2.5).
The concentration of this important cannabis component was reduced to 10% by gamma-irradiation, a technique undertaken to eliminate bacterial contaminants (Hazekamp et al., 2016).
5.2 Caryophyllene Oxide
Caryophyllene oxide is a sesquiterpenoid oxide common to lemon balm (Melissa officinalis), and to the eucalyptus, Melaleuca stypheloides, whose EO contains 43.8% (Farag et al., 2004). Caryophyllene oxide is nontoxic and nonsensitizing, and has the distinction of being the component responsible for cannabis identification by drug-sniffing dogs (Opdyke, 1983; Stahl & Kunde, 1973). This compound serves as a broad-spectrum antifungal in plant defense and as an insecticidal/antifeedant (Bettarini et al., 1993; Langenheim, 1994). Therapeutic applications of caryophyllene oxide could exploit the antifungal efficacy observed in clinical study of onychomycosis compared to ciclopiroxalamine and sulconazole, with an 8% concentration affecting eradication in 15 days (Yang, Michel, Chaumont, & Millet-Clerc, 1999).
This agent also demonstrates antiplatelet aggregation properties in vitro (Lin et al., 2003).
5.3 Humulene (α-Caryophyllene)
Humulene provides some defense to plants and their products, as this compound can inhibit fruit fly mating (Shelly & Nishimoto, 2015). Humulene at a concentration of 1.5 μg/mL produced 50% protection in rat astrocytes against H2O2-induced cell death, and was concentrated seven-fold in those cells (Elmann et al., 2009). The potentiating effect of BCP on the anticancer activity of α-humulene, isocaryophyllene, and paclitaxel against MCF-7, DLD-1, and L-929 human tumor cell lines has been evaluated (Legault & Pichette, 2007). A noncytotoxic concentration of BCP significantly increased the anticancer activity of α-humulene and isocaryophyllene on MCF-7 cells: α-humulene or isocaryophyllene alone (32 μg/mL) inhibited cell growth by about 50% and 69%, respectively, compared with 75% and 90% when combined with 10 μg/mL BCP. Little additional pharmacology, particularly psychopharmacology of the compound has been evident; a recent major review of H. lupulus (hops), of which it is a major EO component, merely mentioned its presence without additional commentary (Zanoli & Zavatti, 2008).
5.4 β-Elemene
Elemene is a monocyclic sesquiterpenoid polyalkene reported from some cannabis chemovars, and common to myrrh (Commiphora myrrha, 9%) and other similar resins (Tisserand & Young, 2014). Elemene via injection has been approved by the regulatory authority in China since 1993 for treatment of cancer. However, a 2006 Cochrane-style review or 127 RCTs showed poor adherence to CONSORT recommendations and very low Jadad scale scoring in available studies (Peng et al., 2006). A subsequent study in rats at
80 mg/kg IV (equivalent to 13 mg/kg in humans) showed good passage through the blood–brain barrier and attainment of high brain tissue levels, as well as noteworthy tumor inhibition and life extension (Wu et al., 2009). A more recent meta-analysis of studies in malignancy (Xu, Zheng, Li, Xu, & Fu, 2013) examined clinical studies up to 2011, examining claims of efficacy in 38 relevant trials. Overall response rate of elemene with chemotherapy was favorable in lung cancer (P<0.00001), hepatic carcinoma (P¼0.002), metastatic brain cancer (P¼0.02), and leukemia (P¼0.0004), but not in gastric carcinoma. Clinical benefit was also seen in combination therapy vs chemotherapy alone in 13 lung cancer trials, 5 with hepatic carcinoma, 7 with gastric carcinoma, and 5 with leukemia, out of 30 examined.
Similar comparison failed to show improved 1-year survival in lung cancer or liver cancer, or 2-year survival in lung cancer.Higher degrees of leukopeniawere significantly lower (P¼0.0007) in the elemene plus chemotherapy groups. Various subsequent studies have examined mechanisms of action of elemene in malignancy. Elemene 100 μM increased cytotoxicity significantly in various cell lines overexpressing the ABCB1 transporter of paclitaxel, colchicine, and vinblastine by inhibiting it efflux activity. Elemene significantly diminished mRNA transcription and P-gp and BCRP gene expression, as well as CD44 and 24-/low cell and CBRP+ cell rates and serum-free cell sphere forming in breast cancer stem cells (Dong et al., 2015). It also dose dependently inhibited survival and proliferation of glioblastoma multiforme cell lines when combined with temozolomide or radiation (Liu et al., 2015) by inhibiting DNA repair via effects on ATM, AKT, and ERK signaling. In A549 human basal cells, elemene increased radiosensitivity through upregulation of p53 and downregulation of Bcl-2-producing apoptosis, and downregulation of DNA-PKcs inhibiting DSB repair (K. Zou, Liu, Zhang, & Zou, 2015). Radiosensitivity of gastric cancer was also enhanced by diminished Pak1 activation (Liu et al., 2015).
Elemene was the first drug reported to inhibit TOPO I and IIα simultaneously, as demonstrated in HepG-2 human hepatic carcinoma, producing cell arrest in S phase and apoptosis (Gong et al., 2015). Elemene mediated multidrug resistance or various genes in exosomes in MCF-7 human breast cancer cells, sensitizing them to docetaxel and adriacin (Zhang et al., 2015). In ECA-109 esophageal carcinoma cells, elemene reduced proliferation significantly via regulation of inhibition of hTERT expression by IncRNA CDKN2B-AS1 (Hu et al., 2015).
In U87 glioblastoma cells, elemene reduced proliferation, increase apoptosis, reduced invasiveness, and mouse xenograft growth (Zhu et al., 2015), while downregulating stemness markers CD133 and ATP-binding cassette subfamily G member 2 and N-cadherin and β-catenin mesenchymal markers. In a review of molecular mechanisms (Jiang et al., 2016), elemene was noted to inhibit cancer growth via multiple mechanisms of proliferative signaling suppression: MAPK and PI3K/Akt/mTOR pathways, upregulation of growth suppressors, promotion of apoptosis, diminishing invasion and metastasis, affecting cell immortality, and reducing angiogenesis.
While concentrations of elemene employed would likely never be attained with cannabis extracts, the distinct possibility of synergy or elemene with chemotherapeutic phytocannabinoids should certainly be explored. Combination with THC, CBD (Marcu et al., 2010), and temozolomide (Torres et al., 2011) for treatment of glioblastoma multiforme would be especially worthy of investigation. A 0.5% elemene emulsion injection proved effective as a sclerosing agent in 23 consecutive patients treated for chylothorax with good reported safety (Jianjun, Song, Yin, Jia, & Donglei, 2008).
Elemene prevented human umbilical vein endothelial cell (HUVEC) damage by hydrogen peroxide in vitro, inhibited smooth muscle proliferation and migration, and neointima formation after vessel injury in rats (Wu, Wang, Tang, Long, & Yin, 2011). In subsequent work (Liu et al., 2015), elemene also decreased reactive oxygen species (ROS) and mitogen-activated protein kinase signaling in HUVECs, and suggesting utility in atherosclerosis treatment. In a rat model of hepatic fibrosis, elemene downregulated plasma endotoxins, serum TNF-α, and expression of CD14, the coreceptor for bacterial lipopolysaccharide detection (Liu et al., 2011).
Elemene 12.5–50 μg/mL inhibited osteogenic differentiation from cultured human hip joint capsule fibroblasts via inhibition of the BMP/SMADs pathway, suggesting its ability to reduce ectopic ossification in ankylosing spondylitis (Zhou et al., 2015). Elemene 10–200 μg/mL also reduced viability and increased apoptosis of rheumatoid arthritis fibroblast-like synoviocytes via induction of ROS and p38 MAPK activation, implying therapeutic potential in that disorder (Zou et al., 2016).
Elemene presence was said to be characteristic of “indica” chemovars of cannabis (Hazekamp et al., 2016). Although its concentrations in most cannabis chemovars are low, emphasis is placed here due to its versatility as a potential anticancer agent worthy of selective breeding to increase its titer, and possibly synergize with chemotherapeutic phytocannabinoids (Ligresti et al., 2006).
5.5 Guaiol
According to Lawless (1995), guaiol, a bicyclic sesquiterpenoid alkene alcohol, is a major component (42%–72%) of the EO of guaiacwood from the species Bulnesia sarmienti, a tree of Paraguay and Argentina, with a pleasant rose-like aroma, and is nontoxic, nonirritating, and nonsensitizing. It has been employed in aromatherapy to treat arthritis, rheumatoid arthritis, and gout. Reported actions of the EO are antiinflammatory, antioxidant, antirheumatic, antiseptic, diaphoretic, diuretic, and laxative. The EO of another species in which guaiol was a component displayed antibiotic properties (de Moura et al., 2002). A report (Parker, 2003) has also shown guaiol to have weak 5-alpha reductase inhibitory effects, and this could be helpful in benign prostatic hyperplasia, or even in treatment of male-pattern baldness, a benefit of cannabis reported independently in the Arabic and Chinese literature.
Guaiol inhibited nonsmall-cell lung cancer cells in vitro, and in vivo in nude mice (as effectively as cisplatin at the same 8 mg/kg dose) (Yang et al., 2016) with mitotic arrest in S phase in A549 and H1299 cells, downregulation of RAD51 homologous recombination repair factor and inducing apoptosis. Guaiol showed contact toxicity for two moth species and efficacy as a fumigant for Musca domestica houseflies with LC50 of 16.9 μL/L (Liu, Wang, Xie, & Mu, 2013). It also demonstrated bite-deterrence index (BDI) against A. aegypti of 0.82, and vs Anopholes quadrimaculatus a BDI of 0.82, comparable to N,N-dimethyl-toluamide (DEET) at a concentration of 25 nM/cm3, but was ineffective against larvae (Ali et al., 2015). Guaiol was said to be a distinguishing factor in Afghan cannabis chemovars (Hillig & Mahlberg, 2004), with similar claim for “indica” chemovars (Hazekamp et al., 2016). As a sesquiterpenoid alcohol, it would be expected to produce sedative effects (Schnaubelt, 1998), often attributed to Afghan genetics.
5.6 Eudesmol Isomers
These isomers are bicyclic sesquiterpenoid alkene alcohols. Presence of both β- and γ-eudesmol isomers was judged to be characteristic of Afghani cannabis (Hillig & Mahlberg, 2004), or “indica” chemovars (Hazekamp et al., 2016). Alpha-eudesmol inhibits calcium channels and was shown to attenuate neurogenic vasodilation, decrease dural extravasation, inhibit depolarizationevoked CGRP and substance P release from sensory nerve terminals without cardiovascular effects (Asakura et al., 2000), suggesting clinical application in migraine.
β-Eudesmol has been reported to be hepatoprotective against carbontetrachloride and galactosoamine-induced cytotoxicity in cultured rat hepatocytes
(Kiso, Tohkin, & Hikino, 1983), and to inhibit electroshock-induced seizures in mice, additive to phenytoin (Chiou, Ling, & Chang, 1997).Other older reports (summarized in Li et al., 2013) note its ability to block nicotinic receptors at the neuromuscular junctions, display antiinflammatory effects, and antagonize toxicity related to organophosphate poisoning.
Recent investigation of β-eudesmol indicates its ability in mice to stimulate gastric emptying and intestinalmotility via inhibition of dopamine D2 and serotonin 5-HT2 receptors in a dose-dependent fashion (25–100 mg/kg) (Kimura & Sumiyoshi, 2012).
Several reports document efficacy in cancer: β-eudesmol produced apoptosis in human leukemia HL60 cell culture, producing apoptosis via effects on JNK signaling in mitochondria (Li et al., 2013); both α- and β-eudesmol produced cytotoxic effects in low μg/mL concentrations in human hepatocellular carcinoma HepG2 cells with increase in caspase-3 activation, loss of mitochondrial membrane potential and apoptosis (D. S. Bomfim et al., 2013); β-eudesmol reduced human cholangiocarcinoma xenograft tumors in nude mice 91.6% at a dose of 100 mg/kg with prolongation of survival by 64.4% (Plengsuriyakarn, Karbwang, & Na-Bangchang, 2015); and it also inhibited proliferation of human lung A549 and colon HT29 and Caco-2 cells, superoxide synthesis in A549, and cell adhesion and migration in A549 and HT29 lines at high concentrations (100 μM) (Ben Sghaier et al., 2016). While it is unlikely that such concentrations would be attainable in herbal cannabis or concentrates, these results suggest possibilities for synergy with other cannabis components.
β-Eudesmol demonstrated BDI against A. aegypti of 0.81, and A. quadrimaculatus mosquitoes with BDI of 0.82, comparable to DEET at a concentration of 25 nM/cm3, but was ineffective against larvae (Ali et al., 2015). Additionally, β-eudesmol in low micromolar concentrations inhibited activity of histidine decarboxylase and mast cell degranulation in a human cell line, HMC-1 (Han et al., 2017), suggesting its possible application in treatment of allergies.
5.7 Nerolidol
Nerolidol, previously reviewed (Russo, 2011), is a noncyclic sesquiterpene alkene alcohol with sedative properties (Binet, Binet, Miocque, Roux, & Bernier, 1972; Lapczynski, Bhatia, Letizia, & Api, 2008), common to citrus peels. It reduced colon adenoma formation in rats (Wattenberg, 1991). It enhanced skin penetration of 5-fluorouracil (Cornwell & Barry, 1994), and it produced growth inhibition of dermophytes (Langenheim, 1994). Unlike some conventional cutaneous preparations, nerolidol is nontoxic and nonsensitizing (Lapczynski, Letizia, & Api, 2008). Potent antimalarial (Lopes et al., 1999; Rodrigues Goulart et al., 2004) and antileishmanial effects (Arruda, D’Alexandri, Katzin, & Uliana, 2005) have been noted, including an IC50 of 7.0 μM in a recent experiment (Camargos et al., 2014). Although present in Sativex®, it seems to exist at only minimal concentration in Californian chemovars (Giese et al., 2015).
5.8 Gurjunene
Gurjunene, a tricyclic sesquiterpene alkene, has also been reported in cannabis, but it is difficult todistinguish it analytically from nerolidol (Hazekamp et al., 2016). Gurjunene is common to EO of agarwood (Aquilaria agallocha) (Takemoto, Ito, Shiraki, Yagura, & Honda, 2008), and when administered as a vapor to mice it produced a biphasic doseresponse, effectively reducing locomotor activity at 1.5%, but stimulating activity at 15% concentration.
5.9 γ-Cadinene
A bicyclic sesquiterpene, while more common in other EOs, cadinene is found in low concentration in current cannabis chemovars tested (Hazekamp et al., 2016). Cadinene demonstrated larvicidal activity against the Anopholes stephensi, vector of malaria (LC50 [lethal concentration] 8.23 μg/mL), A. aegypti, vector of dengue (LC50 9.03 μg/mL), and C. quinquefaciatus, vector of filariasis (LC50 9.86 μg/mL) (Govindarajan, Rajeswary, Hoti, et al., 2016; Govindarajan, Rajeswary, et al., 2016). Little pharmacology of the isolated compound is available, otherwise.
5.10 β-Farnesene
Trans-β-farnesene, an acyclic sesquiterpenes alkene, common to green apple and its scent, and some higher animals, is present in trace amounts in some cannabis chemovars, and was considered characteristic of “sativa” types (Hazekamp et al., 2016). Said to possess DPPH free radical scavenging, anticarcinogenic, antibacterial, and antifungal activity (Turkez et al., 2014), β-farnesene also demonstrated dose-related neuroprotective effects on cultured rat primary cortical neurons, blocking H2O2-induced intracellular LDH release and reduced DNA damage 47.8%, suggesting application in neurodegenerative diseases.
6. CANNABIS ODDS AND ENDS: ROOT TRITERPENOIDS AND ALKALOIDS, LEAF FLAVONOIDS, SEED COATS, AND SPROUTS
While it is clear that the unfertilized female flowering tops with their capitate glandular trichomes are the preeminent phytotherapeutic factory in cannabis, other parts of the plant produce distinctive chemistries of their own, and offer synergistic possibilities in cannabis combinations from this “pharmacological treasure trove” (Mechoulam, 2005). Considering that many parts of the plant are commonly discarded, these “extraneous” materials deserve much closer scrutiny and consideration.
6.1 Friedelin
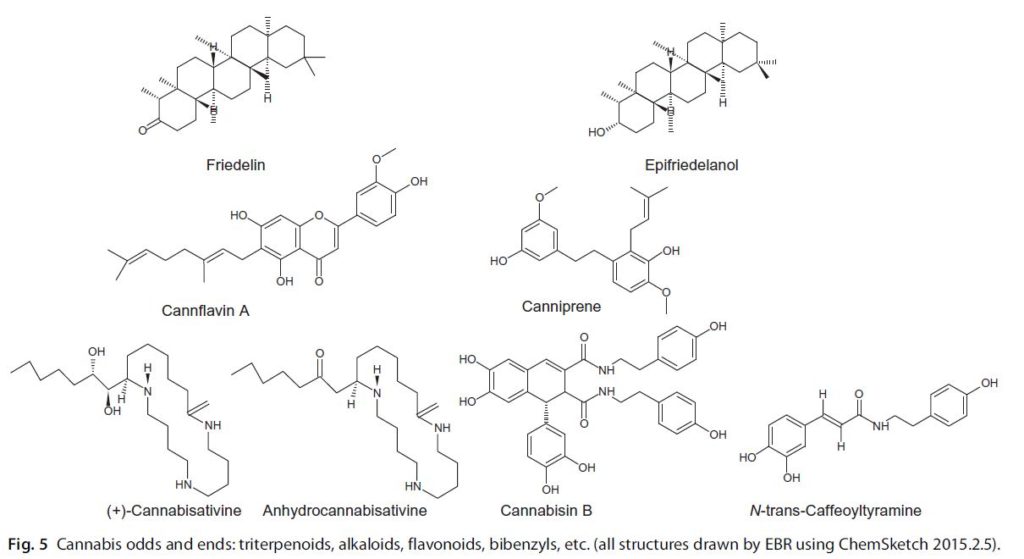
Fig. 5 Cannabis odds and ends: triterpenoids, alkaloids, flavonoids, bibenzyls, etc. (all structures drawn by EBR using ChemSketch 2015.2.5).
While cannabis roots contain no phytocannabinoids (Potter, 2009), monoor sesquiterpenoids, they do produce triterpenoids, the 30-carbon molecules that are the most diverse phytochemicals in nature, derived by cyclization of squalene. They display a wide spectrum of anti-inflammatory, antipyretic, and anticarcinogenic effects with low toxicity (Bishayee, Ahmed, Brankov, & Perloff, 2011). Friedelin is the most prominent triterpenoid in cannabis. It was isolated in 1892 by Friedel, or as early as 1805, as “cerine” and found in many plants (Aesculus, Cannabis, Citrus, Diospyros, Quercus, Rhododendron, Vaccinium), algae, lichen, mosses, peat, coal, mineral wax, and swine (Chandler & Hooper, 1979). Its concentration in cannabis root was calculated as 12.8 mg/kg (Slatkin et al., 1971). Friedelin was weakly active (35 μM or above) against four cancer cell lines (Ee, Lim, Rahmat, & Lee, 2005). More promising were its antiinflammatory and antipyretic effects.
In adult Wistar albino rats, friedelin markedly reduced carrageenan-induced hind paw edema, the effect persisting for 6 h (Antonisamy, Duraipandiyan, & Ignacimuthu, 2011). Results of friedelin at 40 mg/kg dose were comparable to those of indomethacin 10 mg/kg. In the same study, friedelin at doses of 2 or 4 mg markedly reduced rat ear edema after croton oil administration, inhibited peritoneal capillary permeability after acetic acid administration in a dose related manner. Friedelin inhibited granuloma formation after placement of cotton pellets subcutaneously in the axillae, significantly (P<0.05) inhibited paw swelling after Freund’s adjuvant injection, and significantly (P<0.05) reduced abdominal constrictions and stretching after acetic acid injection. The effect was less on first phase (0–5min) neurogenic pain than on second phase (20–30 min) inflammatory pain. Friedelin showed no significant effect vs control on pain threshold in the hot-plate test. Friedelin administered orally showed significant reduction in rectal temperature (P<0.05) after yeast injection, results comparable to the antipyretic effect of paracetamol (acetaminophen).
Additionally, friedelin showed reducing power in vitro, comparable to BHT and ascorbate, which was dose-related (Sunil, Duraipandiyan, Ignacimuthu, & Al-Dhabi, 2013). In five in vitro antioxidant assays, the following results were noted at high concentrations: DPPH radical scavenging effect: IC50 at 21.1 mM, hydroxyl radical scavenging: 50% inhibition at 19.8 mM, nitric oxide radical inhibition: IC50 at 22.1 mM, superoxide radical scavenging: IC50 at 21.9 mM, inhibition of lipid peroxidation: IC50 at 18.1 mM. Friedelin 40 mg/kg pretreatment reduced CCl4-induced LFT elevations due to hepatic damage (P<0.005), comparable to silymarin extract of Silybum marianum (milk thistle). Friedelin 40 mg/kg pretreatment before CCl4 administration produced highly significant increases in superoxide dismutase, catalase, and glutathione peroxidase levels (P<0.005) to normal values, comparable to silymarin. Friedelin demonstrated antimycobacterial activity against three nonpathogenic species at a MIC of 800 μg/mL and merited mention as a natural African antituberculosis agent (Chinsembu, 2016).
Interestingly, this usage parallels that of cannabis leaf macerated in warm water and taken as a treatment for TB by the Bapedi healers of Limpopo Province, South Africa (Semenya, Potgieter, Tshisikhawe, Shava, & Maroyi, 2012), and certainly, friedelin may be contributing to any therapeutic benefit. Friedelin was also effective in protecting against ethanol-induced gastric ulceration in rats (Antonisamy et al., 2015). Oral treatment at 35 mg/kg reduced gross and histological effects, increased mucosal PGE2 level 5.1-fold and NO 2.55-fold, as well as significantly reducing microvascular permeability. Friedelin increased gastric mucus content 3.12-fold and pH 4.03- fold vs control animals, also reducing DNA fragmentation and caspase-3 activity. Overall, the level of gastroprotection with friedelin scored 88.21% as compared to the standard drug, omeprazole, at 90.82%. Synergy of friedelin as a preventive of gastrointestinal ulcers would certainly be possible with reports of similar benefits attributed to THC (Douthwaite, 1947) and caryophyllene (Tambe, Tsujiuchi, Honda, Ikeshiro, & Tanaka, 1996).
6.2 Epifriedelanol
This closely related triterpenoid molecule had a measured concentration in cannabis root of 21.3 mg/kg (Slatkin et al., 1971). It was utilized to assess adriamycin-induced cell senescence in human fibroblasts (HDF) and HUVECs (Yang, Son, Jung, Zheng, & Kim, 2011), wherein epifriedelanol was especially active, and also decreased SA-β-gl activity, p53 protein, and ROS. The authors stated, “This compound [epifriedelanol] may be a promising candidate for developing dietary supplements or cosmetics to modulate tissue aging-associated diseases”.
6.3 Cannabis Root Alkaloids: Cannabisativine and Anhydrocannabisativine
Cannabisativine was isolated from cannabis root (Lotter & Abraham, 1975; Slatkin, Knapp, Schiff, Turner, & Mole, 1975) with calculated concentrations of 2.5 mg/kg (Turner, Hsu, Knapp, Schiff, & Slatkin, 1976) or 0.0004% (Mechoulam et al., 1988). Anhydrocannabisativine was isolated from cannabis roots and leaves (Elsohly et al., 1978), at calculated concentrations of 0.3 mg/kg or 0.00046% (Mechoulam et al., 1988). No pharmacological information is available on either substance: “They are present in miniscule amounts and are presumably not relevant to any cannabis biological activity” (Raphael, Personal comm. to EBR 2013).
6.4 Other Root Components
Slatkin (Slatkin et al., 1975) isolated additional compounds from cannabis root with methanol: sitosterol (calculated content 1.5%), campestrol (0.78%), stigmasterol (0.56%), choline, and neurine. The same author (Slatkin et al., 1971) isolated N(p-hydroxy-β-phenylethyl)-p-hydroxyl-transcinnamamide (1.6 mg/kg), with analgesic activity in the mouse tail flick test at 25, 50, and 100 mg/kg sc.
6.5 Cannabis Seeds
Cannabis or hemp seeds are possibly the single most nutritionally complete food on earth, and reportedly harbor powerful antiinflammatory effects. They contain 35% protein as the digestible edestin, and all essential amino acids. The seeds also contain 35% oil, rich in essential fatty acids in what is considered the ideal nutritional 3:1 ω6:ω3 ratio:75% linoleic acid (LA, ω-6), 25% linolenic acid (LNA, ω-3), and 9% gamma-linolenic acid (GLA, ω-6) (Callaway, 2004). Hemp hulls, often discarded during manufacture of nutritional products, also contain interesting pharmacology. Two compounds were isolated from Chinese varieties (Chen et al., 2012). Antioxidant activity against DPPH radical revealed IC50 9.42 μg/mL for N-trans-caffeoyltyramine and 11.17 μ/mL for cannabisin B, both higher than ariciresinol diglucoside (SDG) and soybean isoflones (ISO). Both compounds also showed prominent
activity in inhibiting human LDL oxidation. In subsequent work (Chen et al., 2013), cannabisin B produced antiproliferative effects in HepG2 human hepatocarcinoma cells (dose dependently up to 500 μM) via arrest in the S phase, and induction of autophagic cell death via regulation of the AKT/mTOR pathway.
6.6 Cannabis Flavonoids
Cannflavin A (CFA) is a flavone unique to cannabis (aerial parts), but is very difficult to isolate and purify via crystallization from its isomer, cannflavin B. It inhibits PGE2 30 times more powerfully than ASA and displays an anti-inflammatory potency intermediate to that of aspirin and dexamethasone (Barrett, Scutt, & Evans, 1986). It remained little studied beyond this for many years, when it was noted to be produced in hemp seed sprouts of certain cultivars (Werz et al., 2014). CFA suppressed PGE2, the primary 114 Ethan B. Russo and Jahan Marcu mediator of inflammation, and directly inhibited mPGES-1 (at 1.8 μM), a target in inflammation and cancer.
Cannflavin A did not significantly inhibit COX-1 nor COX-2, thus potentially avoiding adverse events such as gastrointestinal bleeding, myocardial infarctions, and cerebrovascular accidents associated with the latter agents. The authors indicated that dual inhibition of mPGES-1 and 5-LO is considered the ideal profile to treat inflammatory conditions with fewest side effects. Cannabis leaves contain about 1% total flavonoids, especially apigenin and quercetin (see McPartland & Russo, 2001 for additional review).
6.7 Cannabis Bibenzyl Compounds
Canniprene is an isoprenylated bibenzyl unique to cannabis (Allegrone et al., 2017), that can be vaporized and is potentially present in smoke. Potential anti-inflammatory activity was demonstrated via inhibition of 5-LO (IC50 0.4 μM) and COX/mPGES pathway (IC50 10.1 μM). Related compounds, cannabispiranol and cannabispirenone, were seeming inactive. Canniprene concentration in 160 chemovars ranged trace amounts to more than 0.2% in cannabis leaves. While its concentration did not correlate with phytocannabinoid content or developmental stage of the cannabis plant, it did have a reciprocal relationship with cannflavin A. It will be fascinating to explore the ecological roles that these substances play, and the implications that they might harbor for therapeutic application.
7. CONCLUSION
This review has examined the complex and varied pharmacology of cannabis, a plant that should no longer be considered merely a vehicle for THC, but rather, a potential botanical drug mixture of great therapeutic value in consideration of its genetic plasticity and the promise of its many components. Clinical trials that examine synergistic effects of cannabis components are sorely needed, particularly in the area of phytocannabinoid terpenoid interactions and to assess salient differences between cannabis chemovars attributable to their relative concentrations of entourage compounds.
Contents
1. Introduction
2. Cannabis Phytocannabinoids
2.1 Tetrahydrocannabinol
2.2 Cannabidiol
2.3 Cannabigerol
2.4 Cannabichromene
2.5 Cannabinol
2.6 Tetrahydrocannabivarin
2.7 Tetrahydrocannabinolic Acid
2.8 Cannabidivarin
2.9 Cannabidiolic Acid
2.10 Cannabigerol Monomethyl Ether
3. Cannabis Terpenoids
4. Cannabis Monoterpenoids
4.1 β-Myrcene
4.2 D-Limonene
4.3 β-Ocimene
4.4 γ-Terpinene
4.5 α-Terpinene
4.6 α-Terpineol
4.7 α-Pinene
4.8 β-Pinene
4.9 Linalool
4.10 Camphene
4.11 Terpinolene
4.12 α-Phellandrene
4.13 γ-Cadinene
4.14 Δ3-Carene
4.15 ρ-Cymene
4.16 Fenchol
4.17 1, 8-Cineole (Eucalyptol)
5. Cannabis Sesquiterpenoids
5.1 β-Caryophyllene
5.2 Caryophyllene Oxide
5.3 Humulene (α-Caryophyllene)
5.4 β-Elemene
5.5 Guaiol
5.6 Eudesmol Isomers
5.7 Nerolidol
5.8 Gurjunene
5.9 γ-Cadinene
5.10 β-Farnesene
6. Cannabis Odds and Ends: Root Triterpenoids and Alkaloids, Leaf Flavonoids, Seed Coats, and Sprouts
6.1 Friedelin
6.2 Epifriedelanol
6.3 Cannabis Root Alkaloids: Cannabisativine and Anhydrocannabisativine
6.4 Other Root Components
6.5 Cannabis Seeds
6.6 Cannabis Flavonoids
6.7 Cannabis Bibenzyl Compounds
7. Conclusion